Fast Screening Method for Polymorphisms in Exon 9 of the Catalase Gene.
Abstract
We checked our simple screening technique for detection of the known polymorphism of rs769217, and the two acatalasemic mutations in exon 9 of the catalase gene. This fast and inexpensive method yielded better resolution than those of the standard SSCP. We suppose that the method detects the spontaneously formed single stranded DNAs.
Author Contributions
Academic Editor: Sekhar Kambakam, Iowa State University
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 Laszlo Goth, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
High concentrations of hydrogen peroxide are toxic for human cells and tissues but an increasing body of evidence indicates that its low concentrations function in intracellular signaling 1, 2, 3. The enzyme catalase (EC 1.11.1.6, CAT Gene ID: 847, NM_001752.3, NP_001743.1)4 is the main regulator of hydrogen peroxide metabolism, especially in erythrocytes 5. It decomposes hydrogen peroxide into oxygen and water in a concentration dependent manner. Due to the unique structure of catalase 6, 7 the enzyme decomposes the high (toxic) concentration of hydrogen peroxide while it seems to be unaffected on its low (physiologic) concentration (“flood and gate” mechanism)
Inherited human catalase deficiency(acatalasemia) is associated with disorders8, 9 such as diabetes mellitus10, 11, vitiligo12, dyslipidemia13 and abnormal erythrocyte metabolism14. Fifteen catalase gene (acatalasemic) mutations responsible for decreased catalase activity have been reported from Japan (one missense in exon 4, one splicing in intron 4), North America (one missense in exon 9), Austria (one missense in exon 3) and eleven from Hungary 9, 11.
In exon 9 two acatalasemic mutations were reported one from North America (c. 1093C>T, p.Arg365Cys)15 and two from Hungary (c.1093C>T, p.Arg365Cys and c.1060G>A, p.Arg354His) 8, 9. Furthermore, one silent substitution in exon 9 (rs:769217, c.1058C>T, p.Asp389Asp) was suggested to vitiligo susceptibility12. These reports indicate the increased frequency of catalase gene polymorphisms/mutations in exon 9.
The first detection of rs769214 polymorphism was reported by Wen JK. et al. from Japan 15. They used DNA sequencing while the other authors applied the simpler methods of SSCP 14, and RPLF with BstX1 enzyme 12, 16, 17, 18, 19. The standard version of the single strand conformation polymorphism requires the denaturation (heat, formamide or both) of double strand PCR fragments. This denaturation often leads to the formation of several intermediate strands and sometimes the single strands could not be separated easily by polyacrylamide electrophoresis. The RFLP requires digestion of PCR product by restriction enzyme (BstX1).
Whereas researchers at the next generation sequencing centers have a wide array of core computing resources and expertise, the smaller research and diagnostic laboratories are not so fortunate. To combat the increasing demand from the research and diagnostic fields they are forced to claim and to use simple and cost effective mutation screening methods. Therefore, today’s laboratory professionals are inspired to develop both economical and practical laboratory developed tests to detect DNS polymorphisms which could be used for detection of different diseases.
We have reported on a new and simple method for detection of rs769217 polymorphism in exon 9 of the catalase gene20. In this paper we examined the application of this screening method for detections of polymorphisms and mutations in this exon. We compared the results of this technique to those of standard PCR-SSCP and nucleotide sequencing. We supposed that this method detects single stranded PCR products. The prediction of the secondary single stranded DNA structure was made by Zuker’s method and the effects of the conformational changes on electrophoretic mobilities was also examined.
Patients and Methods
Blood catalase activity was measured by a spectrophotometric method 21 and samples were from the following subjects: D and E types of Hungarian acatalasemia
(n: 4; mean and SD of blood catalase activity 56.7±13.2 MU/L), gestational diabetes (n:43; 78.6±14.0 MU/L), non-insulin dependent diabetes (n:78; 85.3±19.6 MU/L), vitiligo (n:27; 102.4±19.8 MU/L) and controls (n:22; 110.6±15.6 MU/L).
Genomic DNA was extracted from leukocytes using a QIAmp Blood Kit from Qiagen (Hilden, Germany). PCR and primers (5’-TGTTACTGCCCCAGTCAGT-3’ and 5’-ATCTGCTCCACGTGCCCTCT-3’) were the same as described by Kishimoto et al. 22; the region amplified included 139 nucleotides in exon 9, plus 56 nucleotides of intron 8 and 43 nucleotides of intron 9. Reagents (ReadyMix REDTaq with MgCl2) and primers were purchased from Sigma (Sigma-Aldrich, St. Louis, Missouri, USA). Amplifications were performed in total volumes of 12.0 mL. The mixture of 5.0 mL H2O, 1 uL of each primer (10 mmol/L), and 2.0 uL of genomic DNA (0.2 mg/mL) was incubated at 94°C for 5 min. After that 5.0 uL ReadyMix RED Taq (20 mmol/L Tris-HCl pH:8,3, 100 mmol/L KCl, 3 mmol/L MgCl2, 0.002 % gelatine, 0.4 mmol/L dNTP mix, 60 U TaqDNA polymerase) was added. Thirty amplification cycles (94ºC, 55ºC, and 72ºC for 0.5 min, 0.5 min, and 1.0 min, respectively) were performed in a DNA thermal cycler (TC1, Perkin Elmer-Cetus, Norwalk, CT, USA).
For the standard SSCP analyses 5 uL of PCR product and 5 uL of 99 % formamide, 20 mM/L EDTA, 0.05 % bromphenol blue and 0.05 % cylene cyanol were mixed, heated at 96ºC for 6 min and cooled down on ice.
The protocol for the fast SSCP method was reported elsewhere 20. After the PCR 5 µL of its product was mixed with 4 µL of loading dyes (0.05 % bromphenol blue and 0.05 % cylene cyanolin 20 % glycerol) and loaded directly into the polyacrylamide gel. After the electrophoretic separation (6 % polyacrylamide gel at 170 V and room temperature for three hours) the DNA bands were visualized by silver staining.
For heteroduplex formation 2 uL of PCR product was heated to 94ºC, cooled slowly 23 and then mixed with 5 uL of loading dye (0.0125 % bromphenol blue and 0.0125 % cylene cyanolin 20 % glycerol).
For examination of rs 769217 polymorphism in the exon 9 of catalase gene we used the Casp’s RFLP method 12. In this method, BstX 1 enzyme cleaves the 238 bp TT type PCR product into two faster migrating fragments (153bp and 85 bp) but does not cleave the CC type.
Electrophoresis was performed in 6 % polyacrylamide (acrylamide for molecular biology from Sigma-Aldrich and 5xTAE puffer) gel (175x160x1.5 mm) at 170 V and room temperature for three hours. DNA bands were visualized by silver staining and the bands were quantified with Gel Doc 1000 from Bio-Rad.
For DNA sequence analyses, the PCR products were purified by agarose gel electrophoresis. Sequencing reactions were carried out using Taq Dye-Deoxy Termination Cycle Sequencing Kits and DNA fragments were separated and detected by capillary electrophoresis (3100-Avant, Genetic Analyzer, ABI PRISM, Applied Biosystems). Sequencing was performed for all samples in which the screening methods detected polymorphisms. Sequencing reactions were performed in both directions.
Predictions of the secondary structure of DNA utilized the Mfold web server of Zuker 24.
Results and Discussions
For the standard SSCP analyses double stranded PCR products were denatured with formamide and then separated in polyacrylamide gels. Single strand PCR products required strict denaturing conditions and showed only slight differences in their mobilities (Figure 1).
Figure 1.PCR-SSCP pattern of catalase exon 9 with the regular (left) and the new method (right). ss denotes the single stranded bands and ds for the double stranded bands.
The screening method, which was used successfully to detect heteroduplex formation for exon 2 mutations of the catalase gene 23, yielded no detectable heteroduplexes for exon 9 (one band only at molecular mass of 238 bp) for both controls and patients.
For the screening method the incubation at 96˚C for 6 min was omitted. For the standard method it is required for formation of single strands and the fast cooling either for SSCP or slow cooling for heteroduplex formation. Instead of the extra steps of the heating and cooling the PCR products were loaded directly onto the gel with TEA buffer and dyes. After separation of PCR products the silver staining revealed two or more separated bands in the 300-500 bp region (Figure 2). The positions of these bands were similar to the SSCP bands detected using regular PCR-SSCP analyses. Furthermore, these bands were not visualized by ethidium-bromide stain but were detected when the single strand stain SYBR-Green II was used. Quantitation (n=74) revealed that 37±13 % of the silver stained material migrated as 300-500 bp products and 63±15 % migrated like double stranded bands at 240 bp.
Figure 2.The screening PCR-SSCP method yields different migrations for the single stranded SSCP bands (ss) and the double stranded SSCP bands (ds). Lane 1: Mw markers, Lane 2-14 - patients’ samples, rs769217 polymorphism: wild types(CC) in lanes: 2,5,8,10,11, heterozygous (CT) in lanes: 4,7,13 and mutant homozygous (TT) in lanes 3,6,9. c.1060G>A heterozygous mutation in lane 12 and c.1093C>T heterozygous mutation in lane 14.
In the 300-600 bp region (Figure 2) this SSCP method yielded distinct patterns associated with the rs769217 polymorphism as well as the c.1069G>A and c.1093C>T acatalasemic mutations. For the rs769217 polymorphism it yielded different patterns for the TT (lanes 2, 5, 8, 10, 11), TC (lanes 4,7,13) and CC (lanes 3, 6, 9) and two further heterozygote states (lane 12 and 14). Lane 12 (Figure 2) illustrates a heterozygous state with another slow band. Nucleotide analysis revealed a G to A substitution at position 5 of exon 9. This known mutation (c.1060G>A, p.Arg354His) is responsible for Hungarian type D acatalasemia 9. The sample in lane 14 (Figure 2) yielded a heterozygous state with a further intermediate band. Nucleotide analysis revealed a C to T substitution at position 36 of exon 9 (c.1093C>T, p.Arg365Cys). This mutation was found in two sisters who had abnormally low blood catalase activities (71.9 MU/L(65.0%) and 61.9 MU/L(55.9%). Their brother and mother had normal blood catalase activities (110.1 MU/L and 106.3 MU/L); their father had died (Figure 3). This nucleotide substitution causes an Arg to Cys change at amino acid position 365 of catalase. Arg 365, Arg 72 and Arg 112 are important in the neutralization of the charge distribution at Tyr 358, which is essential for the reactivity of catalase’s heme containing active site 6, 7. This nucleotide substitution may be denoted as type E for the Hungarian acatalasemia.
Figure 3.Detection of a Hungarian acatalasemic family with this method. Pedigree (A), SSCP pattern with new method (B), nucleotide sequence analyses of wild type(C) and heterozygote mutant (D).
Figure 4.Evaluation of rs769217 polymorphism in exon 9 of catalase gene with the screening SSCP method in patients with non insulin dependent diabetes mellitus
The Casp’s RFLP method 12 was used to test for T111C polymorphisms in PCR products. Evaluation of samples of 72 subjects with the RFLP 12 and the screening method yielded (Figure 4) the same genetic types for every samples. They were with homozygous wild types (CC: 33), heterozygous types (CT: 31) and homozygous mutant types (TT: 8). These findings were verified in full agreement with those of PCR-DNA sequence analyses(n:4).
No further polymorphisms were detected in the samples of controls (22), and patients with inherited catalase deficiency (4), diabetes mellitus (121), vitiligo (27) 22, 25, 26.
Figure 5.Predicted structures of single strand and loop formatted DNA fragments with different mutations in exon 9 of the catalase gene according to Zuker’ s method24. The predicted structures of the mutant Hungarian type E (upper left) and for Hungarian type D (upper right). The predicted DNA structure of the wild type C (bottom left) of the rs769217 polymorphism and the mutant T type (bottom right). The loops are numbered and arrows show the nucleotide changes.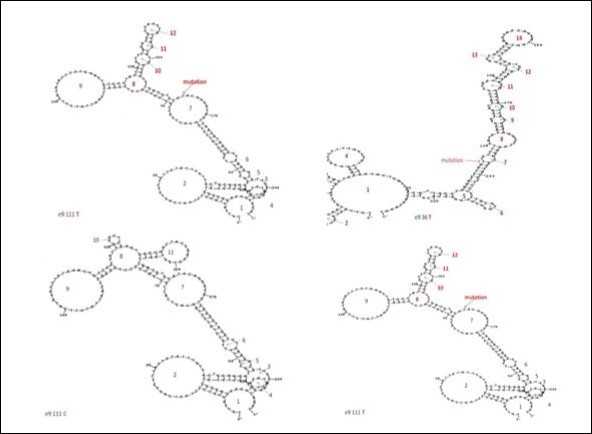
For explanation of the new electrophoretic patterns we used Zuker’s mfold method 24 for prediction of secondary structures in single stranded DNAs, which have loops and double stranded regions (Figure 5and Table 1). We found 14 loops with 6 to 29 nucleotides in the predicted single stranded DNA structures of the wild type and nucleotide substitutions of 111 T, 5 A and 36 C in exon 9. The single stranded DNAs of 111 T, 5A and 36 C have differences from 111 C in number of loops and in size of the loops. The mobility shift between the two strands is the largest for 111 C to T nucleotide substitution while the change in the predicted structure seems to be the small compared to those of the other two nucleotide substitutions.
DNA containing a sequence that generates a local curvature exhibits a pronounced retardation in electrophoretic mobility. Various theoretical models have been proposed but the in silico predictions should be viewed in context of experimental measurement of intrinsic DNA curvature 27. Asingle base substitution or a positional exchange of nucleotide in a highly homologous series of ss-dodecanucleotides led to a change in the mobility-in-gel.Intra-structural transition of single strand DNA based on Watson-Crick base pairing has been reported but little known about its molecular dynamics 28.
For oligomers (16-24 nucleotides) Stellwagen NC. et al. reported on hydrophobic interactions between alkylammonium ions with phosphate residues yielding DNA conformations 29. Their mobility at 20˚C might be due to formation of stable secondary structures in polyacrylamide gels 30. Mobility changes of DNA in polyacrylamide gel might be due to interactions of curved DNAs with polyacrylamide gel matrix 31.
In our case the screening SSCP method revealed single strands (37±13 %) in the 300-800 bp region and double stranded DNA at 240 bp region. The spontaneous formation of single strands (37%) in the new method did not reach the nearly total single strand formation of the regular SSCP. The spontaneously formed single strands are yielding good resolution for detection. Furthermore, their structures are not disturbed either by heating, urea treatment or both.
In accordance to our findings and the former reports 27, 28, 29, 30, 31 we could suggest that some single stranded PCR fragments are prone to internal base pairing. These folding structures may persist and block renaturation of single strands to double strands. Due to the loop formation the conformation of these single stranded DNAs differ from those double stranded DNAs which form base pairs. It yields the slower electrophoretic migration of loop formatted single strand DNA than that of the double stranded DNA.
Beyond, the different migrations of these bands the predicted folded structures showed different dG (minimum free energy 55.7 kcal/mol, 57.3 kcal/mol, 57.4 kcal/mol, 60.0 kcal/mol) and Tm (25. 1°C, 25.5 °C, 25.2 °C, 29.1 °C) values for the wild type and rs769217 (111T, c.1060A, c. 1093C,) mutants. These differences could support the explanation of the different migration rates.
Table 1. Actalasemic mutations in Hungary| Patient | Type | zygote | Gender | Catalase | Exon | Intron | Mutation | ||
|---|---|---|---|---|---|---|---|---|---|
| n | name | MU/L | DNA | Protein | |||||
| 1 | 1 | A | Homozygote | Female | 4% | 2 | 7 | c.138_139insGA | p.Val134X |
| 2 | A | Homozygote | Female | 6% | 2 | c.138_139insGA | p.Val134X | ||
| 3 | A | Heterozygote | Female | 22% | 2 | c.138_139insGA | p.Val134X | ||
| 4 | A | Heterozygote | Female | 46% | 2 | c.138_139insGA | p.Val134X | ||
| 5 | A | Heterozygote | Female | 54% | 2 | c.138_139insGA | p.Val134X | ||
| 6 | A | Heterozygote | Male | 54% | 2 | c.138_139insGA | p.Val134X | ||
| 7 | 2 | B | Heterozygote | Female | 67% | 2 | c.79_80insC | p.Thr71X | |
| 8 | B | Heterozygote | Female | 51% | 2 | c.79_80insC | p.Thr71X | ||
| 9 | B | Heterozygote | Female | 44% | 2 | c.79_80insC | p.Thr71X | ||
| 10 | 3 | C | Heterozygote | Female | 58% | IVS7+5C>T | |||
| 11 | 4 | D | Heterozygote | Female | 45% | 9 | c.1060G>A | p.Arg354His | |
| 12 | 5 | E | Heterozygote | Female | 61% | 9 | c.1093C>T | p.Arg365Cys | |
| 13 | E | Heterozygote | Female | 61% | 9 | c.1093C>T | p.Arg365Cys | ||
| 14 | 6 | F1 | Heterozygote | Female | 53% | 2 | c.161T>A | p.Asp54Glu | |
| 15 | 7 | F2 | Heterozygote | Male | 52% | 2 | c.201G>C | p.Glu68Asp | |
| 16 | 8 | G1 | Heterozygote | Female | 44% | 2 | c.106_107insC | p.Glu71X | |
| 17 | 9 | H1 | Heterozygote | Female | 50% | 4 | c.379C>T | p.Arg127Tyr | |
| 18 | 10 | H2 | Heterozygote | Male | 43% | 4 | c.390T>C | p.Arg130Leu | |
| 19 | 11 | H3 | Heterozygote | Male | 48% | 4 | c.431A>T | p.Asp143Val | |
Conclusions
A simple and inexpensive SSCP method was used for nucleotide mutation screening of exon 9 of the catalase gene. Amplified DNA yielded 37 % single stranded PCR products. They might be due to their changed folding with internal loops which could not form double stranded DNA. These PCR products migrated as distinct bands in the region corresponding to the single stranded DNA region. This screening method is faster and less expensive with a higher resolution than the standard SSCP method. This method verified the known rs769217 polymorphism and the two (c.1060G>A and c.1093C>T) acatalasemic mutations. The application of this method seems to be limited for exon 9 of the catalase gene.
Abbreviations:
TEA: Tris-Acetate-EDTA
SSCP: Single Stranded Conformational Polymorphism
RFLP: Restriction Fragment Length Polymorphism
Contributions:
These authors contributed equally to this work.
References
- 1.Eaton J W. (1999) Hydrogen peroxide a friend of the faux blonde and foe of the cell. , Redox Report 4, 133-134.
- 2.Halliwell B, Veroniqu M, Lung L H. (2001) Hydrogen peroxide in human body. , FEBBS Letts 486, 10-13.
- 3.Veal E, Day A. (2011) Hydrogen peroxide as a signaling molecule. , Antiox. Redox Signal 15, 147-151.
- 5.Mueller S, H D Riedel, Stremmel W. (1997) Direct evidence for catalase as the predominant H2O2 removing enzyme in erythrocytes. , Blood 90, 4973-4978.
- 6.Putnam C D, Arvai A S, Bourne Y. (2000) Active and inhibited human catalase structures: ligand and NADPH binding and catalase mechanism. , J. Mol. Biol 296, 383-393.
- 7.Diaz A, Horjales E, Rudina-Pinera E, Arreola R, Hansberg W. (2004) Unusual Cys-Tyr covalent bond in large catalase. , J. Mol. Biol 342, 295-309.
- 8.Góth L, Rass P, Páy A. (2004) Catalase enzyme mutations and their association with diseases. , Mol. Diagn 8, 141-149.
- 9.Góth L, Nagy T. (2013) Inherited catalase deficiency: Is it a benign factor in various age related diseases?. , Mut.Res./ Rev. Mut. Res 753, 147-154.
- 10.Góth L, Eaton J W. (2000) Hereditary catalase deficiences and increased risk of diabetes. , Lancet 356, 1820-1821.
- 11.Góth L, Nagy T. (2012) Acatalasemia and diabetes mellitus. , Arch. Biochem. Biophys 525, 195-200.
- 12.Casp C B, She J X, McCormack W T. (2002) Genetic association of the catalase gene (CAT) with vitiligo susceptibility. , Pigment Cell Res 15, 62-66.
- 14.Góth L, Vitai M. (2003) The effects of hydrogen peroxide promoted by homocysteine and inherited catalase deficiency on human hypocatalasemic patients. Free Radic. , Biol. Med 35, 882-888.
- 15.Wen J K, Osumi T, Hashimoto T. (1990) Molecular analysis of human acatalasemia. Identification of a splicing mutation. , J. Mol. Biol 211, 383-393.
- 16.Parboosingh J S, Rousseau M, Rogan F, Amit Z, Chetrtkow H et al. (1995) Abscence of mutations in superoxide dismutase and catalase genes in patients with Parkinson’s disease. , Arch. Neurol 52, 1160-1163.
- 17.Park H H, Ha E, Uhm Y K, Jin S Y, Chung J H et al. (2006) Association study between catalase gene polymorphisms and the susceptibility to vitiligo in Korean population. , Exper. Dermatol 15, 377-80.
- 18.Gavalas N G, Akhtar S, Gawkrodger D J, Watson P F, Weetman A P et al. (2006) Analysis of allelic variants in the catalase gene in patients with the skin depigmentation disorder vitiligo. , Biochem. Biophys. Res. Commun 345, 1586-1591.
- 19.Shaji E M, Laddha N C, Chatterjee S, Gani A R, Malek R A et al. (2006) Association of catalase T/C exon 9 and glutathione peroxidase codon 200 polymorphisms in relation to their activities and oxidative stress with vitiligo susceptibility in Gujarat population. , Pigment Cell Res.[Letter] 20, 405-7.
- 20.Nagy T, Góth L. (2012) A simple method for examination of polymorphisms of catalase exon 9: rs769217 in Hungarian microcytic anemia and beta-thalassemia patients. , Arch. Biochem. Biophys 525, 201-206.
- 21.Góth L, Vitai M. (1997) Reference ranges of normal blood catalase activity and levels in hypocatalasemia in Hungary. , Clin. Chim. Acta 261, 35-42.
- 22.Kishimoto Y, Murakami Y, Hayashi K, Takahara S, Sugimura T et al. (1992) Detection of a common mutation of the catalase gene in Japanese acatalasemic patients. , Hum. Genet 88, 487-490.
- 23.Góth L, Gorzsás A, Kalmár T. (2000) A simple PCR-heteroduplex screening method for detection of a common mutation of the catalase gene in Hungary. , Clin. Chem 46, 1199-1200.
- 24.Zuker M. (2003) DNAMelt web server for nucleic acid folding and hybridization prediction. http.//www.bioinfo.rpi.edu//applications/RNA&DNA folding/new version , Nucl. Acid Res 31, 3406-3415.
- 25.Góth L, Csordás M, Kósa Z, Simics E. (2010) A weak association of blood catalase activity and +22348C→T polymorphism of the catalase gene in Hungarian female vitiligo patients. , Clin. Exper. Med.(Budapest) 4, 1-7.
- 26.Tarnai I, Csordás M, Sükei E, Shemirani A H, Káplár M et al. (2007) Effect of C111T polymorphism in exon 9 of the catalase gene on blood catalase activity in different types of diabetes mellitus. , Free Radic. Res 41(7), 806-811.
- 27.Matyáše R, Fulneček J, Kovařík A. (2013) Evaluation of DNA bending models in their capacity to predict electrophoretic migration anomalies of satellite DNA sequences. , Electrophoresis 34(17), 2511-2521.
- 28.Biyani M, Nishigaki K. (2005) Single-strand conformation polymorphism (SSCP) of oligodeoxyribonucleotides: An insight into solution structural dynamics of DNAs provided by gel electrophoresis and molecular dynamics stimulation. , J. Biochem 138, 363-373.
- 29.Chang Y C, Stellwagen N C. (2011) Tandem GA residues on opposite sides of the loop in molecular beacon-like DNA hairpins compact the loop and increase hairpin stability. , Biochem 50(42), 9148-9157.
Cited by (1)
This article has been cited by 1 scholarly work according to:
Citing Articles:
Journal of DNA And RNA Research (2017) OpenAlex