
Molecular Confirmation of Staphylococci Strain’s Identification Isolated in the Hospital and University Center of Brazzaville, Republic of Congo
Abstract
Bacteria of the genus Staphylococcus are pathogenic Gram-positive bacteria responsible for various infections, including skin suppuration, which can be severe or chronic. The objective of this study was to confirm Staphylococci strain’s identification isolated by bacteriological methods from biological products of CHU-B patients, by molecular methods based on the analysis of the gene coding for 16S rRNA. In total, 30 strains of Staphylococci were isolated including 8 (26.66%) community strains, 22 (73.33%) hospital strains. The products of the amplification of gene fragments encoding 16S rRNA from 10 strains of Staphylococci including 6 strains of Staphylococcus aureus (S. aureus) and 4 Coagulase Negative Staphylococci (CNS) were sequenced. The sequences obtained were subjected to bioinformatics analysis to confirm the results of conventional bacteriological methods. Six (6) S. aureus strains, 2 Staphylococcus haemolyticus strains, 1 uncultured bacterium clone nbw618g09c1, and one Staphylococcus sp. have been identified. These results made it possible to confirm the effectiveness of the molecular method and to show the limits of traditional bacteriological methods in the complete identification of bacteria.
Author Contributions
Academic Editor: Jun Wan, Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis IN USA.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2022 Léa Gwladys Gangoue, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Bacteria are micro-organisms or unicellular living beings without a nucleus. Human body harbors several microorganisms, which can be commensal or pathogenic. As soon as they become pathogenic, they can be responsible for many infectious diseases 1 According to the World Health Organization (WHO) bacterial infectious diseases are the 3rd cause of death in developing countries malaria and malnutrition 2. Among the bacteria we cite Staphylococci, which are bacteria responsible for numerous infectious foci which can disseminate and cause septicemia, endocarditis (infection of the endocardium), osteomyelitis (infection of the bone tissue) 3, 4. The species Staphylococcus aureus is the most isolated species and in recent decades has been one of the main causes of hospital or clinical and community infections 5.
Traditional bacteriological methods, namely the study of morphological, cultural and biochemical characters, remain the reference for the identification of the Staphylococcus genus as for all other bacteria. On the other hand, molecular biology techniques are more advantageous for the detection of different bacteria 6,7; and have been able to find their place in routine in a large number of medical analysis laboratories. There is a common molecular biology method for the identification of bacterial strains consisting of bioinformatics analyzes of the nucleotide sequences of the gene encoding 16S ribosomal RNA (16S rRNA), an essential component of the 30S ribosomal complex in prokaryotes, due to conservation of this gene between different species of bacteria. Staphylococcus strain’s identification being an important factor in the management of hospitalized and non-hospitalized infected patients and also a means of overcoming therapeutic failures, this work is therefore part of the aim of confirming by molecular methods the identification of bacteria of the genus Staphylococcus produced by conventional bacteriological methods.
Materials and Methods
Site and Collection of Strains
Staphylococci strains collected at the biomedical analysis laboratory of the Center Hospital and University Center of Brazzaville (CHU-B) using various biological fluids from hospitalized patients (hospital strains) and non-hospitalized patients (community strains) were used.Strains were isolated during the period of April 2020 to July 2020.
Isolation of Strains
The isolation of the strains was carried out on nutrient agar associated with a selective Chapman agar.
Identification of Strains
Classical Bacteriological Methods
The identification of the strains was made on the basis of the morphological characters and the production of a catalase and a staphylocoagulase by the bacterial strain 8.
Molecular Identification
16S rRNA Gene Analysis
Ten strains of Staphylococci identified by conventional bacteriological methods were the subject of a molecular analysis on the analysis of the gene coding for the 16S rRNA.
Extraction of Genomic DNA
DNA extracts were isolated from Staphylococcus strains using the ZR DNA Card extraction Kit while following the manufacturer's instructions.
Amplification of the 16S rRNA Gene
The DNA extracts were amplified by PCR (polymerase chain reaction) using specific primers for the 16S rRNA gene, the universal primers used are those designated by 9 and also used by 10, 11. The sequences and directions of the primers are: Forward F 5’-AGA GTT TGA TCC TGG CTC AG-3 'and Reverse R 5’-ACG GCT ACC TTG TTA CGA CTT-3’.
The PCR reaction was used in a total volume of 50 microliters comprising 39.5 µL of PCR water (Nuclease-free water), 2 µL (20 µM) of each primer, 1 µL (10 µM) of dNTPs, 5 µL of buffer (10X), 2µl (20ng) of genomic DNA and 0.5µL (5Unit/µl) of Taq polymerase. The PCR reaction was carried out in a Biometra thermocycler under the following conditions: pre-denaturation 95°C at 5 min, followed by 30 cycles, each cycle comprising denaturation 95°C at 30 sec, hybridization 55°C, 30 sec and elongation 72° C, 1 min 30s and finally a final elongation 72°C, 5 min.
Agarose Gel Electrophoresis of PCR Product
The PCR products were demonstrated by electrophoresis on a 1% agarose gel at 100 volts for 45 minutes with TBE buffer. The staining was carried out with a 1 μg/ml solution of ethidium bromide. The gel was visualized under a UV lamp, by fluorescence. The size of the sought gene was around 1500-1300 bp.
Sequencing of PCR Products and Assembly of Sequences
The PCR products resulting from the amplification of the gene encoding the 16S rRNA of the ten strains were purified using the NucleoFast 96 PCR plate (Macherey-Nagel EURL, France) and sequenced by the company Masrogen using the BigDye terminator chemistry on an ABI sequencer. 3730 (Applied Biosystems, Foster City, CA, USA). Baser DNA sequence assembler was used for sequence assembly.
In Silico Sequence Analysis
The analysis of the sequences A2, A3, A4, A5, A6, A10 respectively sequences of the strains S2, S3, S4, S5, S6, S10, S. aureus) and A1, A7, A8 A10 of the S1, S7, S8, S10, SCN) was performed using the Local Baseline Alignment Search Tool (BLAST) available at the National Database, Center for Biotechnology Information (http://www. ncbi.nlm.nih.gov). The alignment of the nucleotide sequences was done on ClustalW. Molecular phylogenetic and evolutionary analyzes were performed using MEGA version 7 12.
Results
Isolation and Identification of Strains
Isolation of Strains
Figure 1 represents the percentage of strains of community and clinical Staphylococci isolated and identified. 30 strains of Staphylococci were isolated, including 8 (26.66%) community strains, 22 (73.33%) strains from samples taken from the services in the Hospital and University Center of Brazzaville.
Figure 1.Distribution of isolated strains of Staphylococci.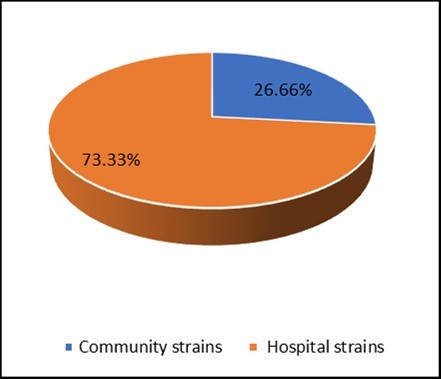
Identification of Strains
All strains isolated were Gram positive and catalase positive bacteria. The fresh state showed that all the stumps were cocci in immobile clusters.
Figure 2 shows the distribution of Staphylococci identified by different coagulase assays. Of the 30 strains of staphylococci isolated from hospitalized and outpatients, 21 (70%) were able to produce a free coagulase (S. aureus), while 9 (30%) were not able to produce it (Coagulase Negative Staphylococcus).
Figure 2.Distribution of strains of Coagulase Negative Staphylococcus and S. aureus identified.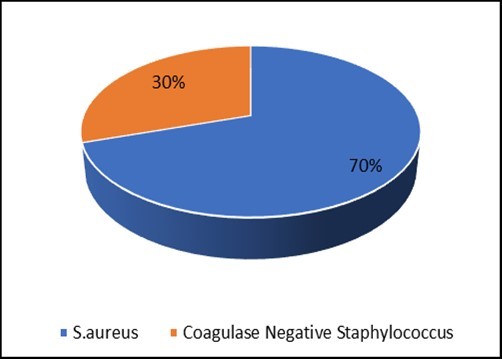
Molecular Identification
Ten strains of clinical Staphylococci, including 6 S. aureus and 4 with Negative Coagulase by classical bacteriological methods, were the subject of a molecular analysis for confirmation on the analysis of the gene coding for 16S rRNA.
DNA extracts from each strain were amplified by PCR using 16S rRNA gene-specific primers.
Electrophoresis of PCR Products
Figure 3 shows the DNA fragments (gene encoding RNA16S) obtained by 1% agarose gel electrophoresis of the 10 strains. The bands are about 1500 Pb in size.
Figure 3.Electrophoresis on Agarose Gel at 1% of the PCR Product of the rRNA16S gene of strains. S1-S10 : Strains of Staphylococci ; M : labder marker DNA (100), CN- : Negative control.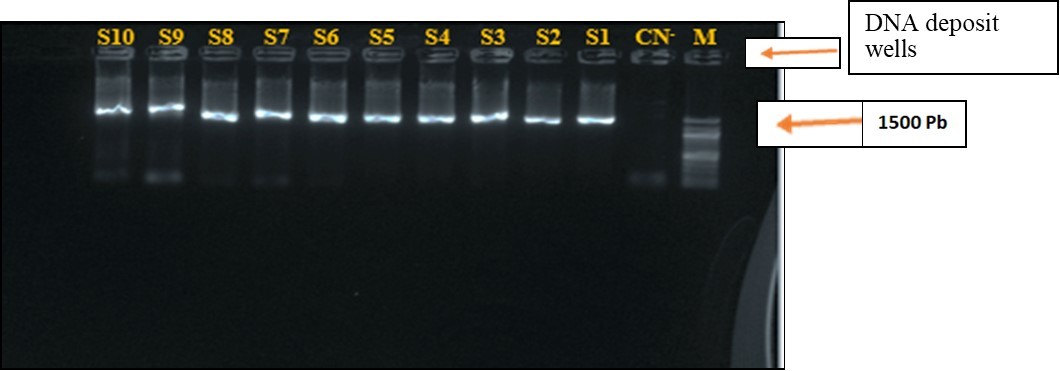
Bioinformatic Analysis of 16S rDNA Sequences and Phylogenetic Inference Test
BLASTN Analysis and Sequences Submission in GenBank
PCR fragments were sequenced and assembled. Table 1 describes the analyzes relating to the similarity of the 16s rRNA sequences between our query sequences and the sequences present in the Gene Bank database, using the bioinformatics algorithm BLASTn. The analysis of this table shows, according to the percentage of similarity, a diversity of species of the identified strains. Table 2
Table 1. Blastn results for strain identification by 16S rRNA gene similarity| Codes | Max Score | E. Value | % de similarité | Accession numbers | Equivalent strains |
| A1 | 1714 | 0.0 | 94,5% | OM281812.1. | Staphylococcus sp. strain PYCC 8255 16S ribosomal |
| A2 | 1725 | 0.0 | 94 % | MW308320.1. | Staphylococcus aureus strain s18 16S ribosomal RNA gene |
| A3 | 1720 | 0.0 | 94 % | MW308321.1. | Staphylococcus aureus strain s18 16S ribosomal RNA gene |
| A4 | 1727 | 0.0 | 91,2% | OP067888.1. | Staphylococcus aureus strain s20 16S ribosomal RNA gene |
| A5 | 1742 | 0.0 | 91,2% | MK809243.1. | Staphylococcus aureus strain RM_AST_SA012 Ribosomal RNA gene 16 |
| A6 | 1705 | 0.0 | 92% | MK809241.1. | Staphylococcus aureus strain RM_AST_SA001 16S ribosomal RNA gene |
| A7 | 1881 | 0.0 | 93,4% | KY218856.1 | Staphylococcus haemolyticus strain AP BFT16 16S ribosomal RNA gene |
| A8 | 1882 | 0.0 | 92 ,4% | MK934564.1. | Staphylococcus haemolyticus strain AP BFT16 16S ribosomal RNA gene |
| A9 | 1714 | 0.0 | 93,5% | GQ110719.1 | Uncultured bacteria clone nbw618g09c1 16S ribosomal RNA gene |
| A10 | 1716 | 0.0 | 94,5 % | AF015929.1. | Staphylococcus aureus 16S ribosomal RNA gene |
| Codes | Sequences of strains identified |
| A1 | Staphylococcus sp. Strain BMGLG21A1 |
| A2 | Staphylococcus aureus Strain BMGLG21A2 |
| A3 | Staphylococcus aureus Strain BMGLG21A3 |
| A4 | Staphylococcus aureus Strain BMGLG21A4 |
| A5 | Staphylococcus aureus Strain BMGLG21A5 |
| A6 | Staphylococcus aureus Strain BMGLG21A6 |
| A7 | Staphylococcus haemolyticus Strain BMGLG21A7 |
| A8 | Staphylococcus haemolyticus Strain BMGLG21A8 |
| A9 | Uncultured bacteria Strain BMGLG21A9 |
| A10 | Staphylococcus aureus Strain BMGLG21A10 |
Multiple Alignment of the Sequences Obtained and Their Homologs in The Databases
Figure 4 represents part of the multiple alignment of the sequences of the RNA16S gene corresponding to all the strains of staphylococci identified and the homologs in the database in particular by using the CLUSTAL W algorithm. These results highlight the highly conserved regions at the level of all the strains due to the T-T-T-T-T-T motif present at the level of all the identified strains, absent at the level of the strain (unidentified clone), as well as mutations (loss of nucleotides).
Figure 4.Part of the multiple alignment of the gene sequences encoding the 16S rRNA of the identified strains and the homologs sequences from the databases.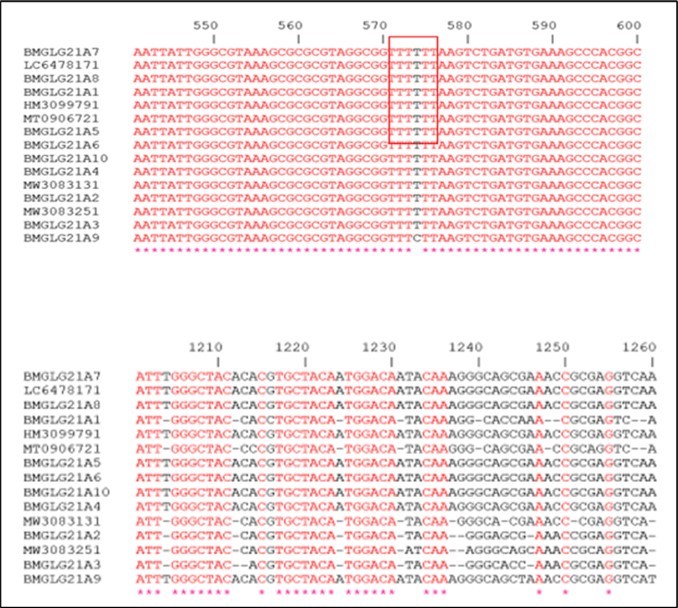
Phylogenetic Classification of the Strains Identified by the Sequencing of 16S rDNA
Figure 5 shows the phylogenetic tree of the identified strains, the species diversity is clearly visible. This tree shows distances of 0.2% (0.002) between strains and that the strains have a common ancestor.
Figure 5.Phylogenetics of strains identified by the 16S rRNA gene and their homologs from databases
The evolutionary history was deduced using the UPGMA method 13. The optimal tree with sum of branch length = 0.05128113 is displayed. The tree is drawn to scale, with branch lengths in the same units as the evolutionary distances used to infer the phylogenetic tree. Evolutionary distances were calculated using the p-distance method 14 and are expressed as the number of baseline differences per site. The analysis focused on 15 nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. All positions with gaps and missing data have been eliminated. There were a total of 1156 positions in the final dataset. Evolutionary analyzes have been carried out in MEGA7 12.
Discussion
Although classical bacteriological methods remain the reference for bacterial identification, we used the molecular method based on the analysis of the gene coding for 16S rRNA for the confirmation of the isolated staphylococcus strains and the different related species identified in this work. Analysis of the results by conventional bacteriological methods show a high percentage of staphylococcus strains from hospital settings (clinical strains) with a prevalence of 67.50%, these results are close to those of 15 and 16. All strains of Staphylococcus were identified as Gram positive and catalase positive bacteria. The results of the fresh state showed Cocci in immobile clusters at the level of all the strains.
1% agarose gel electrophoresis of the strains' 16S rRNA gene PCR products showed a length of approximately 1500 bp. This size (1500Pb) has been endorsed by several studies which have shown the importance of the 16S rRNA gene in the classification and identification of the molecular genus and species of prokaryotes identical to the results obtained by, 17, 18.19. This size indicates that the strains belong to the large group of prokaryotes and certainly to the genus Staphylococcus. Analysis by BLASTn of the sequences of the PCR products, A2, A3, A4, A5, A6, A10 and A7, A8, A9, A10 originating respectively from the strains S2, S3, S4, S5, S6, S10 (S.aureu) s and S7,S8,S9,S6 (SCN) gave E values of 0.0 for all sequences, for each isolate the equivalent based on % similarity of 94.5% with A1 Staphylococcus.sp (OM281812.1 ), A2 94 % S.aureus (MW308320.1), A3 94 % S.aureus (MW308321.1), A4 91.2 % S.aureus (OP067888.1), A5 91.2 % S .aureus (MK809243.1), A6 92 % S. aureus (MK809241.1), A7 93.4 % Staphylococcus haemolyticus, A8 92.4% Staphylococcus haemolyticus (MK934564.1 ,A9 93.5% Bacterium clone uncultivated nbw618g09c1 (GQ110719.1), A10 94.5% S.aureus (AF815929.1.) . The E value as well as the percentage of sequence similarity of the rRNA16S gene of the identified strains show that the results obtained are significant, and make it possible to infer the belonging of the strains, S2, S3, S4, S5, S6, S10 to the species S. aureus, confirming the identification pa r the coagulase test; from S7 and S8 to the Staphylococcus haemolyticus species and the S9 strain to a bacterial clone. The results are different from those presented by 20 during the 15th National Day of Infectiology on the limits and indications of universal PCR of 16S rDNA, indicating the identification of isolates of S. aureus and S. epidermidis at a similarity percentage ˃ 98%. Blast analysis also allowed the identification of two Staphylococcus haemolyticus species and of a bacterial clone of the 4 SCN strains identified by the coagulase test; these results show the limits of traditional bacteriological methods in the complete identification of bacteria.
The analysis of the multiple alignment of the sequences of the RNA16S gene corresponding to all the strains of staphylococci identified and the homologs in the database using the CLUSTAL W algorithm allowed the highlighting of the mutations (losses of nucleotides), These results could explain the low rate of the percentage of similarity observed between the identified sequences and the homologs in Gen Bank. The multiple alignment of the sequences also made it possible to highlight highly conserved regions between the sequences, reflecting the belonging of the identified strains to the same family, to the same genus. These results also show that the strains have undergone evolutionary changes over time, evolutionary changes that are confirmed by the established Phylogenetic tree.
Conclusion
This study made it possible to confirm the identification of Staphylococcus strains made by microbiological methods based on morphological and biochemical characters (coagulase test) by molecular biology techniques, more precisely by the amplification of the 16S rRNA gene from DNA extracts from ten strains. The amplification of the 16S RNA gene is a universal, precise and objective method, whose inter-operator variability is limited compared to conventional techniques, it has enabled us to broaden our knowledge of the bacterial world, more precisely of the genus staphylococcus both at the level of the species and of the genus by highlighting the existing phylogenetic interferences.
References
- 1.Lays. (2012) ARN. Staphylococcus aureus regulators: Role of RsaA in biofilm and capsule formation, RNA expression levels in clinical specimens.
- 3.Johnson AP 2011.Methicillin-resistant Staphylococcusaureus :the European landscape. JAntimicrobChemother66 Suppl 4 : iv43-iv48 .
- 4.Bernier-Lachance J. Quebec. University of Montreal, Faculty of Veterinary Medicine (2015) Prevalence and characterization of methicillin-resistant Staphylococcus aureus of avian origin in. Canada.Master's r memory17p
- 5.Lyon B R, Skurray R. (1987) resistance of Staphylococcus aureus: genetic basis.MicrobiolRev51:. 88-134.
- 6.Drancourt M, Berger P, Raoult D. (2004) . Systematic 16S rRNA Gene Sequencing of Atypical Clinical Isolates Identified 27 New Bacterial Species Associated with Humans.J. ClinMicrobiol: 42(5) : 2197-2202.
- 7.Aurélie Renvoisé 2012. Applicability of "universal" 16S PCR as a bacterial identification and detection tool in the hospital bacteriology laboratory.
- 8.Brown DFJ, Edwards D I, Hawkey P M, Morrison D, Ridgway G L et al. (2005) Joint Working Party of the British Society for Antimicrobial Chemotherapy, Hospital Infection Society. , and Infection Control Nurses Association 56, 1000-1018.
- 9.Weisburg W G, Barns S M, Pelletier D A, Lane D J. (1991) . 16S Ribosomal DNA Amplification for Phylogenetic Study.Journal ofBacteriology 173(2), 697-703.
- 10.Soloka Mabika etal. (2020) Characterization of fibrinolytic enzymes of Bacillus isolated from “Tété” squash and fermented cassava leaves “ntoba mbodi”.Doctoral Thesis46.
- 11.Itsouhou Ngo. (2019) Molecular identification, phylogenetic classification and proteolytic capacity of cultivable bacteria isolated from soils in Brazzaville. , Republic of Congo.JOBIMB 7(2), 1-7.
- 12.Kumar S, Stecher G, Tamura K. (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets.Molecular. , Biology and Evolution 33, 1870-1874.
- 15.Hanane Aouati, Aouati H. Mentouri University. Constantine. Algeria. Magister's Memory (2009) Isolation of Staphylococcus aureus strains resistant to methicillin. Study of their sensitivity to other families of antibiotics. 1-114.
- 16.Tarcisse BALOKI NGOULOU. (2019) Molecular characterization and distribution of genes encoding resistance to Macrolides. Lincosamides and Streptogramins B in community and clinical Staphylococcus in Brazzaville, Congo.Afrique 15(5), 352-363.
- 17.Kim M, Oh H-S, Park S-C, Chun J. (2014) Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes.International. Journal ofSystematicandEvolutionaryMicrobiology64 : 346-351.
- 18.Mohamed M, Saeys Y, Leys N, Raes J, Monsieurs P. (2015) CATCh, an ensemble classifier for chimera detection in 16S rRNA sequencing studies.AppliedandEnvironmentalMicrobiology81 (5) :. 1573-1584.
Cited by (2)
This article has been cited by 2 scholarly works according to:
Citing Articles:
Journal of Biosciences and Medicines (2023) OpenAlex
Journal of Biosciences and Medicines (2023) Crossref