
Abstract
The compound 2-(4-methoxyphenyl)-2, 3-dihydro-1H-perimidine (MPDP) was synthesized. The molecular structure and its functional groups were characterized with the help of Fourier Transform Infrared: FTIR/ Fourier Transform FT-Raman spectra in the regions of 400-4000/50-4000cm-1, respectively. The geometrical parameters, harmonic vibrational wavenumbers, Infrared (IR) & Raman scattering intensities, Nuclear Magnetic Resonance (NMR) chemical shift and Ultraviolet-Visible (UV-Vis) spectra were computed using B3LYP/6-311++G(d,p) level of theory. The complete vibrational analysis were made on the basis of Potential energy distribution (PED) calculation with the help of VEDA4 programme. The Highest occupied molecular orbital (HOMO) – Lowest unoccupied molecular orbital (LUMO) energy gap and intra-molecular charge transfer (ICT) were studied using NBO analysis. The first order hyperpolarizability (β0) and other related properties (β, α0, Δα) of MPDP were computed. The molecular electrostatic potential (MEP), Mulliken atomic charges were calculated using the same level of theory. In addition, the various thermodynamic parameters were also calculated.
Author Contributions
Academic Editor: Zhe-Sheng Chenz, Professor Department of Pharmaceutical Sciences College of Pharmacy and Allied Health Professions St. John's University
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 A Nathiya, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Perimidine derived schiff base have been extensively studied in recent years owing to their great variety of biological activity such as, anti-malarial, antibacterial, anti-tumoral, antiviral activities, etc. 1, 2, 3, 4, 5, 6. Higher π acidity and the presence of more than one hetero atom in pyrimidine play an important role in its co-ordination chemistry, compared to that of pyridine bases, and serve as better models in biological systems 7, 8, 9. Because of perimidine group of organic molecules are reported at scarce. Usually the heterocyclic compounds are used to drug designing in the organic system and the great importance due to possible promising in medical applications 10, 11, 12, 13, 14, 15, 16. Such compounds undergo oxidative aromatization and hydrolysis of the dihydropyrimidine ring. Preparative methods have been developed to facilitate the selective oxidative aromatization or hydrolytic elimination of the dihydropyrimidine fragment of the compounds 17. This kind of compound have been attractive for physicochemical applications of non-linear optical 18, electrical property 19, chelating agents in metal–ligand chemistry as fluorescent liquid crystals 20. Dihydropyrimidinones, the product of the Biginelli reaction, are also widely used in the pharmaceutical industry as calcium channel blockers and alpha-1 antagonists 21. Moreover, some bioactive alkaloids such as batzelladine B, containing the dihydropyrimidine unit, which has been isolated from marine sources, show anti-HIV activity 22.
The biologically relevant molecule 2-(thiophen-2-yl)-2,3-dihydro-1H-perimidine was synthesized and characterized by FT-IR, UV, NMR, MS, CHN microanalysis and X-ray crystallography as well as by DFT/B3LYP/6-311++G(d,p) calculations. The vibrational bands observed in the FTIR spectrum were assigned. The molecular properties like HOMO-LUMO analysis, MEP mapping, chemical reactivity descriptors, dipole moment and natural charges were calculated using the same level of basis set. The results of molecular docking admitted that perimidine may exhibit enzyme inhibitor activity.
In the present study, MPDP molecule was synthesized and characterized by FT-IR, FT-Raman NMR and UV-Vis spectroscopy techniques. The structural and vibrational behavior of the MPDP molecule were carried out using the quantum chemical calculation with the help of DFT/B3LYP/6-311++G (d,p) level of theory.
Experimental Details
Synthesis
The compound MPDP was synthesized by refluxing 4-methoxy benzaldehyde 0.05mol) with 1, 8-diaminonaphthalene 0.05mol) in methanol solution 60ml) for 4 hour. The resulting solution was evaporated in air by slow evaporation to remove the solvent. The product was collected by filtration, washed several time with methanol and recrystallized from hot methanol. The recrystallized product was dried in desiccators under vaccum over anhydrous CaCl2.
FT-IR, FT-Raman, NMR and UV-Vis Spectra Details
The FT-IR spectrum in the spectral range 4000-400 cm-1 was recorded using KBr pellet technique with a FT-IR-Shimadzu spectrometer. The spectrum was recorded at room temperature with a scanning speed of 10 cm-1 per minute at the spectral resolution of 2.0 cm-1 on Instrumentation laboratory, Jamal Mohamed College, Tiruchirappalli, Tamilnadu. FT-Raman spectrum was observed using laser source Nd: YAG 1064 nm as excitation wavelength in the region 50-4000 cm-1 on Bruker IFS 66v spectrophotometer equipped with a FRA 106 FT-Raman module accessory at the spectral resolution of 4 cm-1. The FT-Raman spectrum was recorded at SAIF Laboratory, IIT Madras. The UV-Vis absorption spectrum of MPDP was recorded in the range of 200-500 nm using a Perkin Elmer Lambda-35 spectrometer. The UV pattern was taken from a 10-5 molar solution of MPDP dissolved in methanol, recorded at ACIC, St. Joseph’s College, Tiruchirapalli, Tamilnadu. 1H NMR spectra were recorded on a Bruker 400 MHz spectrometer spectrum is recorded on a Bruker 100 MHz spectrometer in the department of chemistry, Annamalai University, Annamalai Nagar. Chemical shift values are reported in parts per million (ppm) and tetramethylsilane (TMS) as internal standard.
Computational Methods
The entire calculations were performed at DFT levels on a Pentium IV/3.02 GHz personal computer using Gaussian 03W 25 program package, invoking gradient geometry optimization 25, 26. In this study, the density functional three parameter hybrid model DFT/B3LYP/6-311++G (d,p) basis set was used to calculated various properties of the title molecule. The vibrational modes were assigned on the basis of TED analysis with the help of VEDA4 program 27 and by combining results of Gauss View program 28 with symmetry considerations. It should be noted that the Gaussian 03W package is unable to calculate the Raman activity. The Raman activities were transformed into Raman intensities using Raint program 29 by the expression:
Ii =10- 12 x (ν0-νi) 4 x x RAi(1)
Where Ii is the Raman intensity, Ai is the Raman scattering activities, mi is the wavenumber of the normal modes and m0 denotes the wavenumber of the excitation laser 30.
Molecular Docking Study:
Molecular docking experiment was carried out to study the exact binding location of ligand on protein. Molecular docking simulation was performed with the Argus Lab 4.0. The prepared 3D structures of 1JIJ protein was downloaded from the protein data bank (see http://www.rcsb.org/pdb) and binding site was made by choosing “Making binding site for this protein” option. The ligand was then introduced and docking calculation was allowed to run using shape-based search algorithm and A Score scoring function. The scoring function is responsible for evaluating the energy between the ligand and protein target. Flexible docking was allowed by constructing grids over the binding sites of the protein and energy based rotation is set for that ligand group of atoms that do not have rotatable bonds. For each rotation, torsions are created and poses (conformation) are generated during the docking process. For each complex 10 independent runs were conducted and one pose was returned for each run. The best docking model was selected according to the lowest binding energy calculated by Argus lab and the most suitable binding conformation was selected on the basis of hydrogen bond interaction between the ligand and protein near the substrate binding site. The lowest energy poses indicate the highest binding affinity as high energy produces the unstable conformations. The resulting receptor model was saved to Brookhaven PDB file from the file the 2D and 3D interactions are viewed in discovery studio 4.5 versions.
Result and Discussion
NMR Analysis
NMR spectroscopy is a useful technique to establish the structure and nature of the compound. The 1H NMR spectrum of the compound MPDP was recorded in CDCl3 using tetramethyl silane (TMS) as internal standard. 1H NMR spectrum of the compound showed a sharp singlet peak at δ6.839ppm corresponding to C-H proton. A doublet observed at δ9.01ppm with integral value 2 is attributed to >N-H proton in perimidine ring. The multiple signals observed between δ7.196 and 7.836ppm are related to aromatic protons. A sharp peak appeared at δ2.197ppm with an integral value three confirm the presence of –OCH3 group in the compound. Thus 1H NMR results confirmed the formation of MPDP by the condensation of 1, 8, diaminonapthalene and 4-methoxy benzaldehyde. The observed NMR spectral value is listed in Table 1 and the 1H NMR spectrum is shown in Figure 1.
Table 1. The 1H NMR Analysis of MPDP| Compounds | 1H NMR functional groups with chemical shift in δ, ppm) | |||
| -OCH3 | -NH | Aromatic Proton | -HC | |
| Ligand | 2.197 | 4.011, 4.025 | 7.194-7.836 | 6.839 |
Figure 1.The 1H NMR spectrum
Molecular Geometry
The optimized molecular structure of MPDP molecule with atom numbering scheme is shown in Figure 2. The geometrical parameters (Bond lengths, bond angles and dihedral angles) of the title compound was calculated by DFT method and are listed in Table 2. To the best of our knowledge there is no exact crystal data is available for this compound and therefore we have compared the bond parameters with the crystal data of the related molecule.
Figure 2.Optimized structure of 2-(4-methoxyphenyl)-2, 3-dihydro-1H perimidine (MPDP)
| Parameters | B3LYP/ | XRD |
| Bond lengths (Å) | 6-311++G(d,p) | |
| C1-C2 | 1.3842 | 1.366 |
| C1-C6 | 1.4097 | 1.386 |
| C1-H7 | 1.0852 | 0. 930 |
| C2-C3 | 1.4267 | 1.414 |
| C2-N17 | 1.3968 | 1.392 |
| C3-C4 | 1.4265 | 1.412 |
| C3-C8 | 1.4267 | 1.417 |
| C4-C5 | 1.4197 | 1.401 |
| C4-C9 | 1.4197 | 1.401 |
| C5-C6 | 1.3764 | 1.353 |
| C5-H10 | 1.0841 | 0. 930 |
| C6-H11 | 1.0847 | 0. 930 |
| C8-C13 | 1.3842 | 0. 930 |
| C8-N18 | 1.3968 | 1.389 |
| C9-C12 | 1.3764 | 1.345 |
| C9-H14 | 1.0841 | 0. 930 |
| C12-C13 | 1.4097 | 1.398 |
| C12-H15 | 1.0847 | 0. 930 |
| C13-H16 | 1.0852 | 0. 930 |
| N17-H19 | 1.011 | 0.85 |
| N17-C21 | 1.4642 | 1.439 |
| N18-H20 | 1.011 | 0.83 |
| N18-C21 | 1.4642 | 1.461 |
| C21-H22 | 1.1063 | 0. 980 |
| C21-C23 | 1.5112 | |
| C23-C24 | 1.3953 | |
| C23-C25 | 1.3988 | |
| C24-C26 | 1.3937 | |
| C24-H27 | 1.0836 | |
| C25-C28 | 1.3883 | |
| C25-H29 | 1.0855 | |
| C26-C30 | 1.3998 | |
| C26-H31 | 1.0817 | |
| C28-C30 | 1.3993 | |
| C28-H32 | 1.0832 | |
| C30-O33 | 1.363 | |
| O33-C34 | 1.4222 | |
| C34-H35 | 1.0952 | |
| C34-H36 | 1.0886 | |
| C34-H37 | 1.0952 | |
| Bond Angles (°) | ||
| C2-C1-C6 | 120.0186 | 119.7 |
| C2-C1-H7 | 119.9307 | 120.2 |
| C6-C1-H7 | 120.0504 | 120.2 |
| C1-C2-C3 | 119.7025 | 120.1 |
| C1-C2-N17 | 122.7373 | 122.7 |
| C3-C2-N17 | 117.5132 | 117.1 |
| C2-C3-C4 | 120.0261 | 119.6 |
| C2-C3-C8 | 119.9053 | 120.2 |
| C4-C3-C8 | 120.0261 | 120.2 |
| C3-C4-C5 | 118.5802 | 118.2 |
| C3-C4-C9 | 118.5801 | 117.7 |
| C5-C4-C9 | 122.8328 | 124 |
| C4-C5-C6 | 120.2589 | 120.8 |
| C4-C5-H10 | 119.1122 | 119.6 |
| C6-C5-H10 | 120.6271 | 119.6 |
| C1-C6-C5 | 121.4094 | 121.6 |
| C1-C6-H11 | 118.7938 | 119.2 |
| C5-C6-H11 | 119.7963 | 119.2 |
| C3-C8-C13 | 119.7026 | 119.3 |
| C3-C8-N18 | 117.5127 | 117.4 |
| C13-C8-N18 | 122.7376 | 123.2 |
| C4-C9-C12 | 120.2591 | 121.5 |
| C4-C9-H14 | 119.112 | 119.3 |
| C12-C9-H14 | 120.6271 | 119.3 |
| C9-C12-C13 | 121.4094 | 120. 9 |
| C9-C12-H15 | 119.7964 | 119.5 |
| C13-C12-H15 | 118.7938 | 119.5 |
| C8-C13-C12 | 120.0185 | 120.4 |
| C8-C13-H16 | 119.9309 | 119.8 |
| C12-C13-H16 | 120.0503 | 119.8 |
| C2-N17-H19 | 114.7908 | |
| C2-N17-C21 | 116.5255 | 118.3 |
| H21-N17-C19 | 113.1188 | 112 |
| C8-N18-H20 | 114.791 | 113 |
| C8-N18-C21 | 116.5251 | 116.5 |
| C21-N18-H20 | 113.1181 | 113 |
| N17-C21-N18 | 106.5097 | 106.9 |
| N17-C21-H22 | 109.6684 | 109.7 |
| N17-C21-C23 | 111.24 | |
| N18-C21-H22 | 109.6678 | 109.7 |
| N18-C21-C23 | 111.2394 | |
| H22-C21-C23 | 108.4973 | |
| C21-C23-C24 | 120.7247 | |
| C21-C23-C25 | 120.7357 | |
| C24-C23-C25 | 118.5396 | |
| C23-C24-C26 | 121.132 | |
| C23-C24-H27 | 119.1549 | |
| C26-C24-H27 | 119.7131 | |
| C23-C25-C28 | 121.0692 | |
| C23-C25-H29 | 119.6509 | |
| C28-C25-H29 | 119.28 | |
| C24-C26-C30 | 119.6825 | |
| C24-C26-H31 | 119.2783 | |
| C30-C26-H31 | 121.0392 | |
| C25-C28-C30 | 119.9275 | |
| C25-C28-H32 | 121.348 | |
| C30-C28-H32 | 118.7246 | |
| C26-C30-C28 | 119.6493 | |
| C26-C30-O33 | 124.481 | |
| C28-C30-O33 | 115.8697 | |
| C30-O33-C34 | 118.7307 | |
| O33-C34-H35 | 111.4097 | |
| O33-C34-H36 | 105.7893 | |
| O33-C34-H37 | 111.4094 | |
| H35-C34-H36 | 109.3197 | |
| H35-C34-H37 | 109.501 | |
| H36-C34-H37 | 109.3201 | |
| Dihedral Angles (°) | ||
| C6-C1-C2-C3 | 0.111 | |
| C6-C1-C2-N17 | -177.333 | -176.7 |
| H7-C1-C2-C3 | 179.9167 | |
| H7-C1-C2-N17 | 2.4729 | -2.7 |
| C2-C1-C6-C5 | -0.1427 | -0.5 |
| C2-C1-C6-H11 | -179.905 | |
| H7-C1-C6-C5 | -179.948 | -179.8 |
| H7-C1-C6-H11 | 0.2893 | -0.128 |
| C1-C2-C3-C4 | -0.4098 | |
| C1-C2-C3-C8 | -178.038 | |
| N17-C2-C3-C4 | 177.166 | 177 |
| N17-C2-C3-C8 | -0.4624 | 2.4 |
| C1-C2-N17-H19 | -17.688 | 18.5 |
| C1-C2-N17-C21 | -153.242 | 155.7 |
| C3-C2-N17-H19 | 164.8155 | 164 |
| C3-C2-N17-C21 | 29.2611 | 26.8 |
| C2-C3-C4-C5 | 0.7224 | 0.5003 |
| C2-C3-C4-C9 | -178.348 | |
| C8-C3-C4-C5 | 178.3478 | |
| C8-C3-C4-C9 | -0.7229 | |
| C2-C3-C8-C13 | 178.0387 | -179.8 |
| C2-C3-C8-N18 | 0.4635 | 2.4 |
| C4-C3-C8-C13 | 0.4104 | -0.3 |
| C4-C3-C8-N18 | -177.165 | |
| C3-C4-C5-C6 | -0.7531 | -0.0028 |
| C3-C4-C5-H10 | 179.7372 | -179. 9 |
| C9-C4-C5-C6 | 178.2757 | -179.6 |
| C9-C4-C5-H10 | -1.234 | -0.304 |
| C3-C4-C9-C12 | 0.7534 | 0.003 |
| C3-C4-C9-H14 | -179.737 | -179. 9 |
| C5-C4-C9-C12 | -178.276 | 179.6 |
| C5-C4-C9-H14 | 1.2341 | -0.304 |
| C4-C5-C6-C1 | 0.4711 | -0.2 |
| C4-C5-C6-H11 | -179.769 | 179.8 |
| H10-C5-C6-C1 | 179.9732 | 179.8 |
| H10-C5-C6-H11 | -0.2666 | 0.12 |
| C3-C8-C13-C12 | -0.1114 | 0.58 |
| C3-C8-C13-H16 | -179.917 | -179.89 |
| N18-C8-C13-C12 | 177.3319 | -176.7 |
| N18-C8-C13-H16 | -2.4735 | 2.7 |
| C3-C8-N18-H20 | -164.816 | -164 |
| C3-C8-N18-C21 | -29.263 | -26.8 |
| C13-C8-N18-H20 | 17.6879 | -18.5 |
| C13-C8-N18-C21 | 153.241 | -155.7 |
| C4-C9-C12-C13 | -0.471 | 0.2 |
| C4-C9-C12-H15 | 179.769 | 179.8 |
| H14-C9-C12-C13 | -179.973 | -179.8 |
| H14-C9-C12-H15 | 0.2669 | |
| C9-C12-C13-C8 | 0.1427 | |
| C9-C12-C13-H16 | 179.9478 | 179. 9 |
| H15-C12-C13-C8 | 179.905 | 179.8 |
| H15-C12-C13-H16 | -0.2898 | 0.26 |
| C2-N17-C21-N18 | -54.1289 | 47.1 |
| C2-N17-C21-H22 | 64.4735 | |
| C2-N17-C21-C23 | -175.5 | |
| H19-N17-C21-N18 | 169.5971 | -175 |
| H19-N17-C21-H22 | -71.8004 | |
| H19-N17-C21-C23 | 48.2258 | |
| C8-N18-C21-N17 | 54.13 | |
| C8-N18-C21-H22 | -64.4728 | |
| C8-N18-C21-C23 | 175.5018 | |
| H20-N18-C21-N17 | -169.597 | |
| H20-N18-C21-H22 | 71.8002 | |
| H20-N18-C21-C23 | -48.2252 | |
| N17-C21-C23-C24 | 59.278 | |
| N17-C21-C23-C25 | -120.723 | |
| N18-C21-C23-C24 | -59.2881 | |
| N18-C21-C23-C25 | 120.7114 | |
| H22-C21-C23-C24 | 179.9955 | |
| H22-C21-C23-C25 | -0.005 | |
| C21-C23-C24-C26 | 179.9994 | |
| C21-C23-C24-H27 | -0.0003 | |
| C25-C23-C24-C26 | -0.0001 | |
| C25-C23-C24-H27 | -180 | |
| C21-C23-C25-C28 | -179.999 | |
| C21-C23-C25-H29 | 0.0009 | |
| C24-C23-C25-C28 | 0.0008 | |
| C24-C23-C25-H29 | 180.0005 | |
| C23-C24-C26-C30 | -0.0007 | |
| C23-C24-C26-H31 | 179.9992 | |
| H27-C24-C26-C30 | 179.9991 | |
| H27-C24-C26-H31 | -0.0011 | |
| C23-C25-C28-C30 | -0.0007 | |
| C23-C25-C28-H32 | 179.9997 | |
| H29-C25-C28-C30 | 179.9997 | |
| H29-C25-C28-H32 | 0.0001 | |
| C24-C26-C30-C28 | 0.0008 | |
| C24-C26-C30-O33 | -180 | |
| H31-C26-C30-C28 | 180.001 | |
| H31-C26-C30-O33 | -0.0003 | |
| C25-C28-C30-C26 | -0.0002 | |
| C25-C28-C30-O33 | 180.001 | |
| H32-C28-C30-C26 | -180.001 | |
| H32-C28-C30-O33 | 0.0006 | |
| C26-C30-O33-C34 | -0.0008 | |
| C28-C30-O33-C34 | -180.002 | |
| C30-O33-C34-H35 | 61.3032 | |
| C30-O33-C34-H36 | 179.9994 | |
| C30-O33-C34-H37 | -61.304 |
It is evident from the Table 2, that the deviations are observed in case of C-H, and N-H bond length while other bond length are in agreement with the literature 23. The calculated C-H bond length of perimidine ring (except:C21-H22) is deviated slightly ( ̴ 0.1547Å) from the literature value 23, which may be arise from the low, scattering factors of hydrogen atoms in the x-ray diffraction 23. Normally the C-H bond distance is observed around 1.0840Å, whereas the calculated bond length of C21-H22 is 1.063Å. This negative ( ̴ 0.02 Å) deviation is due to the existence of twisted boat configuration caused by the methoxy substitution at C21 atom. The bond C21-C23 has lesser bond length (1.5112Å) among the other C-C bonds. These may be due to the attachment of methoxyphenyl with C21 atom.
In perimidine ring, the various bond angles are found to be consistent with literature value 23. The bond angles of C5-C4-C9/C2-C3-C8 are calculated as 122.83˚/119. 91˚, respectively and are differ by 2.92˚. This observation confirmed the involvement of C2-C3-C8 carbon chain in of perimidine ring structure. The calculated bond angles C25C28H32/C24C26H3 and C30C28H32/C30C26H31 are found at 121.35˚/119.28˚ and 118.73˚/121.04˚, respectively. Furthermore, the bond angles C28C30O33 (115.87˚) and C26C30O33 (124.48˚) are differ by ( ̴ 8.61˚). These observations suggest the distortion of the bulky methoxy group.
The dihedral angle of C3-C2-N17-C21 is 29.26 and C3-C8-N18-C21 is -29.26. This dihedral angle indicated that N17-C21-N18 ring portion projected in the upward direction to attain the twisted boat configuration. This twisted boat configuration is attained due to the steric effect of methoxy group substituted at C21 atom.
Vibrational Assignments
The MPDP molecule pocess C1 point group symmetry. It consists of 37 atoms, and hence 105 normal modes of vibrations are possible and are also distributed as: 71 in-plane bending and 34 out-of-plane bending vibrations. The detailed vibrational assignment are made on the basis of PED analysis using VEDA4 program 31. The calculated frequencies are usually higher than the corresponding experimental quantities, due to the combination of electron correlation effects and basis set deficiencies. These discrepancies were overcome by applying the appropriate scaling factors; the theoretical calculations reproduce the experimental data well in agreement. The calculated wavenumbers scaled with a proper scale factor 32. The simulated and observed FT-IR and FT-Raman spectra are shown in Figure 3, Figure 4, respectively. The experimental and scaled theoretical harmonic vibrational frequencies along with the calculated PED values are given in Table 3.
Figure 3.Combined experimental and theoretical FT-IR spectra of MPDP
Figure 4.Combined experimental and theoretical FT-Raman spectra of MPDP
| Mode No. | Calculated | Observed | IR | Raman | Reduced | Force | Vibrational Assignments≥10% (PED) d | ||
| Frequencies (cm -1 ) | Frequencies (cm -1 ) | Intensity | Intensity | Masses | Consts. | ||||
| Un Scaled | Scaled a | FT-IR | FT-Raman | Rel. b | Rel. c | ||||
| 1 | 3586 | 3445 | 6.85 | 0.44 | 1.08 | 8.15 | νN17H19(100) | ||
| 2 | 3586 | 3445 | 3357 w | 3244 w | 4.56 | 2.15 | 1.08 | 8.16 | νN17H19(100) |
| 3 | 3207 | 3081 | 3076 w | 3080 w | 3.06 | 1.75 | 1.09 | 6.62 | νC26H31(99) |
| 4 | 3194 | 3068 | 2.19 | 2.75 | 1.09 | 6.57 | νC28H32(93) | ||
| 5 | 3186 | 3061 | 2.11 | 0.48 | 1.09 | 6.51 | νC24H27(99) | ||
| 6 | 3184 | 3059 | 11.89 | 8.14 | 1.1 | 6.55 | νC1H7(98) | ||
| 7 | 3182 | 3057 | 12.36 | 1.18 | 1.1 | 6.54 | νC1H7(99) | ||
| 8 | 3168 | 3044 | 3043 w | 4.64 | 3.37 | 1.09 | 6.44 | νC5H10(98) | |
| 9 | 3167 | 3043 | 6.5 | 0.02 | 1.09 | 6.44 | νC1H7(99) | ||
| 10 | 3161 | 3037 | 4.09 | 0.92 | 1.09 | 6.41 | νC25H29(94) | ||
| 11 | 3156 | 3032 | 8.66 | 0.18 | 1.09 | 6.37 | νC1H7(98) | ||
| 12 | 3156 | 3032 | 0.05 | 1.97 | 1.09 | 6.37 | νC1H7(99) | ||
| 13 | 3136 | 3013 | 3013 w | 10.68 | 2.79 | 1.1 | 6.37 | νC34H36(91) | |
| 14 | 3066 | 2946 | 2934 w | 15.64 | 1.02 | 1.11 | 6.13 | νC34H35(99) | |
| 15 | 3007 | 2889 | 2839 w | 25.69 | 3.45 | 1.03 | 5.51 | νC34H37(91) | |
| 16 | 2889 | 2776 | 35.44 | 2 | 1.08 | 5.32 | νC21H22(100) | ||
| 17 | 1660 | 1595 | 18.65 | 0.29 | 6.14 | 9.96 | βH7C1C2(46)+νC1C2(23) | ||
| 18 | 1651 | 1586 | 1589 m | 1591 m | 18.2 | 3.93 | 5.83 | 9.37 | νC21N18(74)+βH27C24C26(19) |
| 19 | 1634 | 1570 | 188.95 | 14.61 | 5.8 | 9.12 | νC12C13(55) | ||
| 20 | 1626 | 1562 | 17.72 | 11.46 | 5.95 | 9.26 | νC1C6(76)+βH10C5C4(18) | ||
| 21 | 1617 | 1554 | 1544 w | 19.53 | 1.32 | 6.55 | 10.09 | νC23C24(68)+βC24C26C30(26) | |
| 22 | 1549 | 1489 | 1512 s | 0.01 | 0.13 | 3.8 | 5.37 | νC1C6(49)+νC1C2(26)+βH7C1C2(20)+νC1C6(24) | |
| 23 | 1544 | 1484 | 47.62 | 0.94 | 2.55 | 3.58 | βH27C24C26(23)+νC21C23(20)+νC26C30(32) | ||
| 24 | 1517 | 1457 | 1464 m | 52.21 | 11.23 | 1.68 | 2.28 | βH10C5C4(58) | |
| 25 | 1504 | 1445 | 24.61 | 1.22 | 1.06 | 1.42 | βH35C34H37(68)+τH35C34O33C30(25) | ||
| 26 | 1493 | 1435 | 4.12 | 1.29 | 1.05 | 1.37 | βH35C34H36(66)+ΓC34H35O33H36(26) | ||
| 27 | 1492 | 1433 | 1428 w | 1.26 | 0.71 | 2.69 | 3.53 | νC1C2(36)+βH7C1C2(33) | |
| 28 | 1476 | 1418 | 3.07 | 0.34 | 1.19 | 1.53 | βH35C34H36(81) | ||
| 29 | 1468 | 1410 | 0.5 | 0.07 | 1.64 | 2.09 | βH19N17C2(52)+τH22C21C23C24(32) | ||
| 30 | 1461 | 1404 | 12.21 | 2.07 | 2.16 | 2.71 | νC24C26(33)+βH7C1C2(21)+βH32C28C30(20)+βH10C5C4(25) | ||
| 31 | 1447 | 1391 | 17.46 | 1.38 | 2.21 | 2.73 | νC12C13(38)+βH10C5C4(55) | ||
| 32 | 1435 | 1379 | 1365 w | 1374 w | 100 | 2.4 | 3.74 | 4.53 | νC24C26(23)+νC1C2(33) |
| 33 | 1401 | 1346 | 15.73 | 13.79 | 5.07 | 5.86 | νC2C3(33)+νC3C4(24) | ||
| 34 | 1385 | 1331 | 0.46 | 3.96 | 2.08 | 2.35 | νC2C3(18)+βH7C1C2(40) | ||
| 35 | 1349 | 1296 | 1.19 | 0.05 | 2.39 | 2.56 | βH7C1C2(25)+νC1C2(28)+τH22C21C23C24(32) | ||
| 36 | 1333 | 1280 | 1280 w | 10.8 | 1.25 | 2.85 | 2.98 | νC23C24(21)+βH7C1C2(46) | |
| 37 | 1322 | 1271 | 1271 s | 6.85 | 2.5 | 1.81 | 1.86 | νC23C24(20)+βH7C1C2(56) | |
| 38 | 1316 | 1265 | 4.99 | 0.71 | 1.23 | 1.26 | βH19N17C2(12)+τH22C21C23C24(42) | ||
| 39 | 1285 | 1235 | 52.56 | 6.63 | 1.57 | 1.52 | νC12C13(38)+βH7C1C2(51) | ||
| 40 | 1278 | 1228 | 127.37 | 2.41 | 4.21 | 4.05 | νO33C30(59) | ||
| 41 | 1268 | 1219 | 1212 m | 6.75 | 1.24 | 2.84 | 2.69 | τH22C21C23C24(32)+νN17C2(15)+νC4C5(33)+νC1C6(24) | |
| 42 | 1234 | 1185 | 1190 w | 0.1 | 0.16 | 1.87 | 1.68 | νC1C2(36)+βH7C1C2(53) | |
| 43 | 1229 | 1181 | 0.95 | 11.48 | 2.3 | 2.05 | βH27C24C26(15)+νC21C23(70) | ||
| 44 | 1207 | 1159 | 0.05 | 0.08 | 1.28 | 1.1 | νC1C6(26)+βH10C5C4(68) | ||
| 45 | 1201 | 1154 | 5.52 | 0.86 | 1.42 | 1.21 | βH35C34H37(38)+τH35C34O33C30(45) | ||
| 46 | 1190 | 1144 | 40.16 | 2.62 | 1.17 | 0.98 | νC23C24(24)+βH27C24C26(29)+νC26C30(32) | ||
| 47 | 1190 | 1143 | 4.21 | 0.29 | 1.37 | 1.14 | νC1C2(18)+βH7C1C2(51) | ||
| 48 | 1168 | 1122 | 0.27 | 0.34 | 1.27 | 1.02 | βH35C34H36(16)+ΓC34H35O33H36(46) | ||
| 49 | 1147 | 1102 | 4.12 | 1.18 | 1.92 | 1.49 | νC1C6(25)+βH7C1C2(36) | ||
| 50 | 1136 | 1092 | 8.02 | 0.63 | 3.96 | 3.01 | νC1C6(74) | ||
| 51 | 1133 | 1088 | 3.1 | 0.05 | 1.31 | 0.99 | νC24C26(23)+βH32C28C30(61) | ||
| 52 | 1109 | 1065 | 1077 w | 15.4 | 1.57 | 1.95 | 1.41 | βH10C5C4(25)+νC1C2(63) | |
| 53 | 1094 | 1051 | 10.03 | 6.42 | 2.39 | 1.69 | νC1C6(29)+νN17C21(56) | ||
| 54 | 1058 | 1017 | 1030 m | 30.98 | 0.76 | 6.72 | 4.43 | νO33C34(72)+νC26C30(12) | |
| 55 | 1054 | 1012 | 3.72 | 0.28 | 3.43 | 2.24 | νC4C5(40)+βC3C2N17(18) | ||
| 56 | 1026 | 986 | 1.11 | 0.1 | 2.63 | 1.63 | βH27C24C26(43)+τH19N17C2C1(14) | ||
| 57 | 995 | 956 | 2.29 | 1.59 | 4.41 | 2.57 | νN18C21(76) | ||
| 58 | 976 | 938 | 0.04 | 0.02 | 1.34 | 0.75 | τH27C24C26C30(34)+τC8C13C9C12(41) | ||
| 59 | 973 | 935 | 0.24 | 0.24 | 1.29 | 0.72 | τH7C1C2N17(38)+βC8C13C12(34) | ||
| 60 | 967 | 929 | 0.01 | 0.01 | 1.29 | 0.71 | τH7C1C2N17(61) | ||
| 61 | 960 | 922 | 0.12 | 0 | 1.35 | 0.73 | τH27C24C26C30(17)+ΓC21C23C25C24(33) | ||
| 62 | 886 | 851 | 863 w | 2.2 | 2.74 | 3.95 | 1.83 | νN18C8(53) | |
| 63 | 875 | 840 | 0.34 | 0.49 | 1.55 | 0.7 | τH7C1C2N17(51) | ||
| 64 | 861 | 827 | 7.06 | 0.01 | 3 | 1.31 | βC4C9C12(28)+τH27C24C26C30(32)+βC3C2N17(18) | ||
| 65 | 855 | 821 | 822 w | 0.12 | 0.22 | 1.43 | 0.62 | τH7C1C2N17(52) | |
| 66 | 842 | 809 | 15.26 | 0.03 | 2.07 | 0.86 | βC4C9C12(28)+τH27C24C26C30(22)+βC21C23C25(27) | ||
| 67 | 826 | 794 | 797 w | 25.76 | 0.86 | 4.08 | 1.64 | τH7C1C2N17(21)+τC8C13C9C12(35)+βC28C30O33(26) | |
| 68 | 825 | 793 | 1.79 | 0.05 | 1.29 | 0.52 | τH27C24C26C30(61) | ||
| 69 | 818 | 786 | 789 w | 13.52 | 3.67 | 4.56 | 1.8 | νC1C2(53)+βC2C1C6(28) | |
| 70 | 792 | 761 | 756 w | 0.13 | 3.25 | 4.64 | 1.71 | νO33C30(59)+νC26C30(22) | |
| 71 | 776 | 745 | 42.78 | 0.31 | 5.16 | 1.83 | βC2C1C6(38)+βN17C21N18(32) | ||
| 72 | 767 | 737 | 34.36 | 0.02 | 1.57 | 0.54 | τH7C1C2N17(21)+βC28C30O33(46) | ||
| 73 | 755 | 725 | 0.03 | 0.13 | 1.31 | 0.44 | τH7C1C2N17(22)+ τH10C5C4C9(70) | ||
| 74 | 744 | 715 | 701 w | 0.36 | 0.54 | 3.67 | 1.2 | τH27C24C26C30(27)+τC8C13C9C12(30) | |
| 75 | 671 | 645 | 639 w | 2.39 | 12.72 | 5.04 | 1.34 | νN18C8(33)+νC2C3(44) | |
| 76 | 651 | 626 | 0.23 | 0.08 | 3.05 | 0.76 | ΓC1C3N17C2(29) | ||
| 77 | 650 | 624 | 8.14 | 1.28 | 5.48 | 1.36 | βC24C26C30(26)+τC8C13C9C12(45) | ||
| 78 | 641 | 616 | 0.17 | 1.47 | 4.81 | 1.17 | βC24C26C30(16)+τC8C13C9C12(25)+βC28C30O33(16)+ΓC8C2C4C3(22) | ||
| 79 | 620 | 596 | 4.13 | 0.28 | 2.68 | 0.61 | βC4C9C12(18)+ΓC8C2C4C3(15)+τC9C4C12C13(25)+βC3C2N17(18) | ||
| 80 | 594 | 570 | 11.71 | 0.45 | 2.3 | 0.48 | βC23C21N18(21)+τH27C24C26C30(22)+τC9C4C12C13(25)+τC8C13C9C12(30) | ||
| 81 | 574 | 551 | 558 w | 17.35 | 8.87 | 5.88 | 1.14 | νC2C3(44)+βC8N18C21(36) | |
| 82 | 546 | 524 | 85.88 | 3.72 | 1.35 | 0.24 | ΓC5C3C9C4(61) | ||
| 83 | 532 | 511 | 0.45 | 0.48 | 2.85 | 0.48 | τC9C4C12C13(25)+τH19N17C2C1(16)+ΓO33C26C28C30(27)+βC21C23C25(17) | ||
| 84 | 521 | 501 | 1.88 | 2.74 | 4.93 | 0.79 | βC3C2N17(18)+βC8N18C21(36) | ||
| 85 | 507 | 487 | 494 w | 0.12 | 1.07 | 3.55 | 0.54 | τH19N17C2C1(26)+τC25C28C26C30(30)+ΓO33C26C28C30(27) | |
| 86 | 495 | 476 | 0.75 | 0.6 | 4.76 | 0.69 | τH19N17C2C1(36)+βC21C23C25(47) | ||
| 87 | 473 | 455 | 459 w | 0.83 | 0.04 | 3.44 | 0.45 | βC8C13C12(44)+ΓC8C2C4C3(32) | |
| 88 | 464 | 446 | 448 m | 0.18 | 0.59 | 4.87 | 0.62 | ΓC1C3N17C2(45) | |
| 89 | 432 | 415 | 2.18 | 0.7 | 4.35 | 0.48 | νN18C8(23)+νN17C21(16)+νC1C2(13)+βN17C21N18(22) | ||
| 90 | 429 | 413 | 0.01 | 0.6 | 3.09 | 0.34 | τH27C24C26C30(24)+τC8C13C9C12(31) | ||
| 91 | 416 | 399 | 0.05 | 6.56 | 3.16 | 0.32 | τC9C4C12C13(35)+ΓC1C3N17C2(52) | ||
| 92 | 361 | 347 | 0.16 | 0.2 | 5.45 | 0.42 | βC26C30C28(57) | ||
| 93 | 323 | 310 | 1.34 | 3.23 | 3.98 | 0.25 | βC24C26C30(42)+βC28C30O33(39) | ||
| 94 | 261 | 251 | 2.42 | 0.79 | 3.72 | 0.15 | βC24C26C30(72) | ||
| 95 | 257 | 247 | 0 | 0.4 | 3.47 | 0.13 | τC25C28C26C30(67) | ||
| 96 | 238 | 228 | 0.21 | 0.68 | 1.27 | 0.04 | βC3C2N17(14) | ||
| 97 | 230 | 221 | 1.15 | 2.89 | 6.41 | 0.2 | νC1C2(33)+βC24C26C30(42) | ||
| 98 | 172 | 166 | 3.05 | 1.51 | 4.4 | 0.08 | τC6C1C2N17(49) | ||
| 99 | 170 | 164 | 0.39 | 0.77 | 3.11 | 0.05 | βC23C21N18(21)+βC21C23C25(27)+τC6C1C2N17(10)+τC2N17N18C21(13) | ||
| 100 | 144 | 138 | 0.43 | 3.9 | 4.55 | 0.06 | βC28C30O33(19)+τC23C25C30C28(29)+ΓC8C2C4C3(22) | ||
| 101 | 142 | 136 | 0.23 | 0.81 | 4.29 | 0.05 | τC2N17N18C21(58) | ||
| 102 | 95 | 91 | 1.52 | 2.4 | 3.59 | 0.02 | βC23C21N18(31)+βC26C30C28(27)+τC6C1C2N17(10) | ||
| 103 | 42 | 41 | 0.05 | 31.32 | 5.17 | 0.01 | βC28C30O33(19)+τC23C25C30C28(29)+τH35C34O33C30(31) | ||
| 104 | 40 | 38 | 0.1 | 19.16 | 4.59 | 0 | βC23C21N18(61)+τC2N17N18C21(33) | ||
| 105 | 29 | 27 | 0.31 | 100 | 3.7 | 0 | ΓC1C3N17C2(57) | ||
N-H Vibrations
Zhuomin Li et al., 33 assigned the νNH, βNH and ΓNH at 3367, 1278 and 544cm-1, respectively in the case of 1’,3’-dihydrospiro fluorene. The stretching and in-plane bending modes of NH group of perimidine ring in 2-(thiophene-2yl)-2,3-dihydro-1H-perimidine (TDP) were respectively assigned at 3413 and 1516cm-1 in FTIR spectrum 23. The IR absorption bands due to νNH mode usually occur in the region of 3200-3500cm-134. The βNH vibration is an important mode, which shows their presence as medium to strong bands in the region 1549-1520cm-134.
In the present molecule the observed vibrational frequencies at 3357/3244 cm-1 in FT-IR/ FT-Raman spectra and its corresponding calculated values are at 3445/3445 cm-1(mode no: 1& 2) are assigned to νN–H mode. These modes are in expected regions and also find support from PED value (100%). The observed frequencies are negatively ( ̴ 88cm-1) deviated from harmonic frequencies, which is due to the presence of moderate unharmonicity in this vibration as well as may be the participation of NH bond in the inter-molecular hydrogen-bonding interactions occur with in the molecule/crystal 23. The N–H in-plane bending modes (βH19N17C2 are assigned at 1410 and 1265cm-1 in MPDP molecule (mode nos: 29 & 38). The N-H torsional mode τH19N17C2C1 is assigned at 494 cm-1 in FT-IR spectrum and for the same mode the corresponding harmonic values are 487, 476cm-1 (mode nos:85, 86) harmonic values. The above assignments are consistent with literature 23 and also find support from PED values 52, 12 and 26, 36%).
C-H Vibrations:
The bands vibrating at 2935, 2851cm-1 in FTIR spectrum were ascribed to C-H stretching vibrations of perimidine ring in TDP by M. Alum et al., 23. The scaled harmonic frequencies in the range 3059-3032cm-1 (mode nos:6-9, 11, 12) are attributed to νC-H modes of perimidine ring MPDP. It should be mentioned here that these νC-H modes are pure and having >98% of PED value.
In aromatic compounds, the νC-H, βC-H and ΓC-H bending modes appear in the ranges of 3100-3000, 1300-1000 and 1000-750cm-1, respectively 35. In this study, there are eleven C-H stretching βC-H and ΓC-H vibrations are possible for MPDP]. As revealed by PED, the phenyl ring νC-H modes are undoubtedly assigned at 3081, 3068, 3061 and 3037cm-1 (mode nos: 3-5, 10). The observed weak bands at 3076/3080cm-1 (FTIR/FT-Raman) are in agreement with mode no: 3. The theoretically calculated values of C-H in-plane bending vibrations of phenyl ring vibrations fall in the region 1404-986cm-1 (mode nos:30, 46, 51, 56). Similarly the harmonic bands due to C-H out-of-plane bending vibration fall in the region 938-715cm-1 (mode nos:58, 64, 68, 74). In this study, the experimental bands are missing for βCH mode, which may be due to the steric factor of bulky group (naphthalene ring). The βCH and τCH modes are within the expected region with few exceptions and also having considerable PED values: >20 and 27%, respectively. The observed FTIR band at 701cm-1 is further support the mode no:74. Literature survey reveal that the CH group shows characteristic frequency band due to stretching/in-plane bending vibrations are in the regions of 2975-2840/1470-1370 cm-1, respectively23.
M. Alam et al., 23 assigned the βCH/τC-H modes in the ranges of 1549-1180cm-1/906-773cm-1, respectively for TDP molecule. In this study, based on the above conclusion the harmonic bands 1595, 1489, 1457, 1391, 1271, 1185 mode nos:17, 22, 24, 31, 37, 42) with considerable PED value( >20%) are assigned to βC-H modes. The mode nos:22, 24, 37, 42 are in moderate agreement with literature value 23 in addition to experimental values: 1512, 1464, 1271 (FTIR), 1190cm-1 (FT-Raman). Similarly the calculated value of τC-H modes of perimidine ring are: 935, 840, 821, 794, 737, 725cm-1 (mode nos: 59, 63, 65, 67, 72, 73), in which mode nos:65, 67 are agreement with observed spectral values (822:FTIR, 797cm-1:FT-Raman). These assignments also having considerable PED (>20%) value. Besides the νC21-H22, βC21-H22 and τC21-H22 deformation vibrations are attributed to mode nos: 16, and 38. On comparing νC-H, these assignments with literature value (νC-H: 2935cm-1, βC-H: 1340) is negatively ̴ 159cm-1 deviated. This is due to attachment of o-methoxy phenyl with C21 atom.
O–CH3 Group Vibrations:
In methoxy group the C–H stretching bands were observed at 2977, 2828, 2838 cm−1/2971, 2929, 2838 cm−1 in FT-IR/FT-Raman spectra are assigned to C–H asymmetric/ symmetric stretching modes, respectively 36. The stretching mode of O–CH3 was assigned in the region of 1000–1100 cm−1 for anisole and its derivatives by some workers 37, 38, 39, 40. In this study, the O–CH3 stretching mode observed at 1030/FT-IR cm−1 and its corresponding calculated value is 1017 cm−1 (mode no: 54). This assignment is further supported by PED value (72%).
Literature survey 37, 40 revealed that the C-O-CH3 angle bending mode have been assigned in the region of 300-670cm-1 for anisole and its derivatives. 36 assigned this mode at 344cm-1 (FT-Raman) in the case of 4-methoxy-2-methyl benzoic acid. In accordance with above facts, the band observed at 494 cm-1 in FTIR and its corresponding harmonic value 487 cm-1 (mode no:85) is assigned to C-O-CH3 angle bending mode. This mode lies in the region of ring planar C-C-C angle bending modes. In this study, a strong mixing of τC25C28C26C30 mode with the C-O-CH3 angle bending mode. This trend is supported by literature 36.
Meganathan et al 36 assigned the torsional mode of the O–CH3 group at 54 cm-1 in FT-Raman spectrum. In this study, the harmonic value 41 cm-1/mode no: 103 assigned to the O–CH3 torsional mode. These assignments are having considerable PED values (27, 31%).
C-N Vibrations:
Generally the νC=N/νC-N vibrations were observed in the regions of 1600–1670 cm-1 /1266-1382cm-134, 41. According to the work by Z.Li and W. Deng 23 on 1’,3’-dihydrospiro fluorine, the harmonic/observed wavenumber 1611/1623cm-1 : FTIR are assigned to νC-N mode. The C=N (aromatic) stretching mode appeared in the region 1490-1570 cm-1 khalaji et al., 2010 42.
The C-N stretching vibrations are assigned to harmonic wavenumbers :1586, 1219, 956, 851cm-1 (mode nos:18, 41, 57, 62), in which mode nos:18, 41, 62 are in agreement with the observed FTIR bands:1589 (1591: FT-Raman), 1212, 863 cm-1. These assignments are supported by their PED values. Besides, the observed FTIR bands: 1497, 1287cm-1 were assigned to νC-N mode for TDP 23. The assigned νC-N modes are negatively/positively deviated from the literature values, which is due to the possibility of C-N bond polarization in the present molecule.
The in-plane bending vibrations: βC3C2N17; βN17C21N18 and βC8N18C21 are attributed to mode nos:64, 79; 71, 89, and 81, 84 respectively. The out-of-plane bending mode of ΓC-N contributes as mixed vibrations of ΓC1C3N17C2 (mode nos: 88, 91). While comparing, the recorded values for mode nos: 81/88 lie at 558/448cm-1 (FTIR bands) and 551/446cm-1 as harmonic frequencies which contribute about 45/52% of PED.
C–O Vibrations:
In C-O group, the absorption is sensitive for both the carbon and oxygen atoms. Normally the C–O stretching vibration occurs in the region 1000–1260 cm-136. Meganathan et al., 2010 have assigned at 1251 cm-1 (FTIR) and 1248 cm-1 (FT-Raman) to aforementioned band. The intensity of the carbonyl group increases, due to the conjugation (or) formation of hydrogen bonds. The increase in conjugation, which increase the intensity of Raman lines as well as the IR band intensities 43. According to the above facts, the harmonic frequencies 1228, 1017 (mode nos:40, 54) are designated as νC30-O33, νC34-O33 modes, respectively with considerable PED values (59, 72%) as well as relative IR intensities (127.37, 30. 98). The mode no:54 is further supported by observed FTIR band: 1030cm-1 with medium intensity.
The in-plane bending vibrations of βC-O are observed as mixed vibration of βC28C30O33 at 797 cm-1 in FT-Raman spectrum, whereas the calculated frequencies are: 794, 737 (mode nos: 67, 72). Similarly, the C-O out-of-plane bending modes are also contribute as mixed vibration of ΓO33C26C28C30 at 494cm-1 in FT-IR and the harmonic frequencies are:511, 487 (mode nos: 83, 85). These assignments are having >26% of PED and also find support from literature 36.
C-C Vibrations:
The carbon–carbon (C-C) stretching vibrations were reported in the regions of 1625–1590, 1590–1575, 1540–1470, 1465–1430 and 1380–1280 cm-1 with variable intensity by Varsanyi 44. In this study, the bands observed at 1544, 1365, 1271 cm-1 in FTIR and at 1280 in FT-Raman spectra are attributed to νC-C mode for phenyl ring in MPDP and their corresponding harmonic frequencies are: 1554, 1484, 1404, 1379, 1280, 1271 (mode nos :21, 23, 30, 32, 36, 37). The PED value corresponding to all νC-C modes lie in between 20 and 68% and also mixed with βCH mode. The ring breathing, in-plane and out-of-plane bending modes belong to phenyl ring were assigned at 726 (FT-Raman), 694 and 235cm-1 in FTIR spectrum, respectively 36. The observed FTIR band at 756cm-1 (harmonic: 761cm-1/mode no:70) is assigned to ring breathing mode and moderately in agreement with above literature value. Similarly, the theoretically calculated frequencies 624, 616 and 487, 247 (mode nos: 77, 78 and 85, 95) are belong to βCCC and ΓCCC modes, respectively. These assignments are further supported by PED value ( >16%).
Alam and Lee 23 assigned the stretching vibrations of C-C in the wavenumber range 1180-1771cm-1 (FTIR) for TDP. The theoretically calculated νC-C modes:1562-1065cm-1 (mode nos:20, 22, 27, 31-33, 39, 41, 42, 50, 52) have been obtained to be consistent with the recorded spectral values:1512, 1428, 1365, 1212 cm-1 (FTIR bands) and 1190, 1077cm-1 (FT-Raman). The βCCC and τCCC modes were assigned at 796, 522 and 773, 471cm-1, respectively for TDP 23. Based on the above literature value, the harmonic frequencies in the ranges of 827-455cm-1 (64, 66, 69, 71, 79, 87) and 616-138cm-1 (78, 79, 82, 87, 100) are assigned to βCCC and ΓCCC modes of dyhydro perimidine ring, respectively. These assignments are in agreement with literature 23 and also having considerable PED values. (>33%: νCC; >18%: βCCC; >15%: ΓCCC). The νC21C23, βC21C23C25 and ΓC21C23C25C24 modes are assigned to mode nos: 43, 66 and 61, respectively. According to PED results the βCCC and ΓCCC vibrations are mixed with βCH mode.
Nonlinear Optical Effects (NLO)
The NLO materials have recently attracted much interest because they involve new scientific phenomena and also they offer potential applications in emerging optoelectronic technologies, telecommunications, information storage, optical switching and signal processing 45. The output from GAUSSIAN 03W provides 10 components of the 3x3x3 matrix as βxxx; βxxy; βxyy; βyyy; βxxz; βxyz; βyyz; βxzz; βyzz; βzzz; respectively. The total static dipole moment (µ), polarizability (α0), and the first hyperpolarizability (β0) can be calculated by using the following equations,
The dipole moment, polarizability and the first hyperpolarizability were calculated using B3LYP/6-311++G(d,p) basis set. As can be seen from Table 4, the calculated values of electronic dipole moment (µ), polarizability (α0), and the first hyperpolarizability (β0) are 1.3385 Debye, 0.6255x10-30 esu and 3.0847x10-30 esu, respectively. The first order hyper polarizability (β0) value is 18 times greater than that of urea and hence the molecule has good NLO property.
Table 4. The NLO measurements of MPDP| Parameters | B3LYP/6-311++G(d,p) |
| Dipole moment ( μ ) Debye | |
| μx | -1.3379 |
| μy | 0.00007 |
| μz | -0.0385 |
| μ | 1.3385Debye |
| Polarizability ( α0 ) x10-30esu | |
| αxx | 343.81 |
| αxy | 0.00 |
| αyy | 228.13 |
| αxz | -0.72 |
| αyz | -0.00 |
| αzz | 160.95 |
| α0 | 0.62559x10-30esu |
| Hyperpolarizability ( β0 ) x10-30esu | |
| βxxx | 366.33 |
| βxxy | 0.01 |
| βxyy | -44.56 |
| βyyy | 0.02 |
| βxxz | 100.48 |
| βxyz | 0.00 |
| βyyz | -14.30 |
| βxzz | 1.72 |
| βyzz | -0.01 |
| βzzz | 64.94 |
| β0 | 3.08470x10-30esu |
NBO Analysis:
The non-covalent bonding and anti-bonding interaction can be quantitatively described in terms of the NBO analysis, which is expressed by means of the second-order perturbation interaction energy (E(2)) 46, 47, 48, 49, 47, 48, 49, 50. This energy represents the estimation of the off-diagonal NBO Fock matrix elements. It can be deduced from the second-order perturbation approach 50
where qi is the donor orbital occupancy, Σi and Σj are diagonal elements (orbital energies) and F(i, j) is off diagonal NBO Fock matrix elements.
NBO analysis was performed by the B3LYP/6-311++G(d,p) basis set for the molecule MPDP and are listed in Table 5. The intramolecular hyperconjugative interactions energes are formed by the orbital overlap between π(C–C) and π*(C–C) bond orbitals, which results intra molecular charge transfer (ICT) causing stabilization of the system.
Table 5. The second order perturbation theory analysis of Fock Matrix in NBO basis for MPDP| Type | Donor NBO (i) | ED/e | Acceptor NBO (j) | ED/e | E(2)KJ/mol | E(j)-E(i) a.u. | F(i,j)a.u. |
| σ -σ* | BD ( 1) C 1 - C 2 | 1.97745 | BD*( 1) C 1 - C 6 | 0.01374 | 10.5 | 1.27 | 0.05 |
| BD*( 1) C 1 - H 7 | 0.01199 | 5.61 | 1.19 | 0.036 | |||
| BD*( 1) C 2 - C 3 | 0.02874 | 17.66 | 1.28 | 0.066 | |||
| BD*( 1) C 3 - C 8 | 0.02874 | 12.84 | 1.28 | 0.056 | |||
| BD*( 1) C 6 - H 11 | 0.01105 | 8.45 | 1.2 | 0.044 | |||
| BD*( 1) N 17 - C 21 | 0.02799 | 5.73 | 1.03 | 0.034 | |||
| π -π* | BD ( 2) C 1 - C 2 | 1.70554 | BD*( 2) C 3 - C 4 | 0.5074 | 63.18 | 0.3 | 0.063 |
| BD*( 2) C 5 - C 6 | 0.29421 | 88.99 | 0.29 | 0.07 | |||
| σ -σ* | BD ( 1) C 1 - C 6 | 1.97803 | BD*( 1) C 1 - C 2 | 0.01907 | 12.22 | 1.26 | 0.054 |
| BD*( 1) C 1 - H 7 | 0.01199 | 5.86 | 1.18 | 0.036 | |||
| BD*( 1) C 2 - N 17 | 0.02361 | 17.74 | 1.04 | 0.059 | |||
| BD*( 1) C 5 - C 6 | 0.01359 | 9.83 | 1.26 | 0.049 | |||
| BD*( 1) C 5 - H 10 | 0.01155 | 9.41 | 1.19 | 0.046 | |||
| BD*( 1) C 6 - H 11 | 0.01105 | 4.77 | 1.19 | 0.033 | |||
| σ -σ* | BD ( 1) C 1 - H 7 | 1.98106 | BD*( 1) C 1 - C 2 | 0.01907 | 4.35 | 1.1 | 0.03 |
| BD*( 1) C 2 - C 3 | 0.02874 | 17.82 | 1.11 | 0.062 | |||
| BD*( 1) C 5 - C 6 | 0.01359 | 13.85 | 1.1 | 0.054 | |||
| σ -σ* | BD ( 1) C 2 - C 3 | 1.96808 | BD*( 1) C 1 - C 2 | 0.01907 | 14.94 | 1.27 | 0.06 |
| BD*( 1) C 1 - H 7 | 0.01199 | 8.24 | 1.19 | 0.043 | |||
| BD*( 1) C 3 - C 4 | 0.03301 | 19.04 | 1.29 | 0.068 | |||
| BD*( 1) C 3 - C 8 | 0.02874 | 17.49 | 1.28 | 0.065 | |||
| BD*( 1) C 4 - C 9 | 0.0215 | 10.46 | 1.28 | 0.051 | |||
| BD*( 1) C 8 - C 13 | 0.01908 | 10.71 | 1.27 | 0.051 | |||
| BD*( 1) N 17 - H 19 | 0.01237 | 4.39 | 1.18 | 0.032 | |||
| σ -σ* | BD ( 1) C 2 - N 17 | 1.98308 | BD*( 1) C 1 - C 6 | 0.01374 | 6.74 | 1.29 | 0.041 |
| BD*( 1) C 2 - C 3 | 0.02874 | 4.23 | 1.3 | 0.032 | |||
| BD*( 1) C 3 - C 4 | 0.03301 | 9.79 | 1.31 | 0.05 | |||
| BD*( 1) C 21 - C 23 | 0.03162 | 8.16 | 1.11 | 0.042 | |||
| σ -σ* | BD ( 1) C 3 - C 4 | 1.95847 | BD*( 1) C 2 - C 3 | 0.02874 | 19.54 | 1.26 | 0.069 |
| BD*( 1) C 2 - N 17 | 0.02361 | 15.23 | 1.04 | 0.055 | |||
| BD*( 1) C 3 - C 8 | 0.02874 | 19.54 | 1.26 | 0.069 | |||
| BD*( 1) C 4 - C 5 | 0.0215 | 14.56 | 1.26 | 0.06 | |||
| BD*( 1) C 4 - C 9 | 0.0215 | 14.56 | 1.26 | 0.06 | |||
| BD*( 1) C 5 - H 10 | 0.01155 | 7.87 | 1.19 | 0.043 | |||
| BD*( 1) C 8 - N 18 | 0.02365 | 15.23 | 1.04 | 0.055 | |||
| BD*( 1) C 9 - H 14 | 0.01154 | 7.87 | 1.19 | 0.043 | |||
| π -π* | BD ( 2) C 3 - C 4 | 1.5483 | BD*( 2) C 1 - C 2 | 0.3377 | 95.1 | 0.27 | 0.072 |
| BD*( 2) C 5 - C 6 | 0.29421 | 63.55 | 0.28 | 0.06 | |||
| BD*( 2) C 8 - C 13 | 0.33793 | 95.23 | 0.27 | 0.072 | |||
| BD*( 2) C 9 - C 12 | 0.29421 | 63.64 | 0.28 | 0.06 | |||
| σ -σ* | BD ( 1) C 3 - C 8 | 1.96807 | BD*( 1) C 1 - C 2 | 0.01907 | 10.71 | 1.27 | 0.051 |
| BD*( 1) C 2 - C 3 | 0.02874 | 17.45 | 1.28 | 0.065 | |||
| BD*( 1) C 3 - C 4 | 0.03301 | 19.04 | 1.29 | 0.068 | |||
| BD*( 1) C 4 - C 5 | 0.0215 | 10.46 | 1.28 | 0.051 | |||
| BD*( 1) C 8 - C 13 | 0.01908 | 14.94 | 1.27 | 0.06 | |||
| BD*( 1) C 13 - H 16 | 0.01201 | 8.24 | 1.19 | 0.043 | |||
| BD*( 1) N 18 - H 20 | 0.0124 | 4.39 | 1.18 | 0.032 | |||
| σ -σ* | BD ( 1) C 1 - C 2 | 1.97745 | BD*( 1) C 1 - C 6 | 0.01374 | 10.5 | 1.27 | 0.05 |
| BD*( 1) C 1 - H 7 | 0.01199 | 5.61 | 1.19 | 0.036 | |||
| BD*( 1) C 2 - C 3 | 0.02874 | 17.66 | 1.28 | 0.066 | |||
| BD*( 1) C 3 - C 8 | 0.02874 | 12.84 | 1.28 | 0.056 | |||
| BD*( 1) C 6 - H 11 | 0.01105 | 8.45 | 1.2 | 0.044 | |||
| BD*( 1) N 17 - C 21 | 0.02799 | 5.73 | 1.03 | 0.034 | |||
| π -π* | BD ( 2) C 1 - C 2 | 1.70554 | BD*( 2) C 3 - C 4 | 0.5074 | 63.18 | 0.3 | 0.063 |
| BD*( 2) C 5 - C 6 | 0.29421 | 88.99 | 0.29 | 0.07 | |||
| σ -σ* | BD ( 1) C 1 - C 6 | 1.97803 | BD*( 1) C 1 - C 2 | 0.01907 | 12.22 | 1.26 | 0.054 |
| BD*( 1) C 1 - H 7 | 0.01199 | 5.86 | 1.18 | 0.036 | |||
| BD*( 1) C 2 - N 17 | 0.02361 | 17.74 | 1.04 | 0.059 | |||
| BD*( 1) C 5 - C 6 | 0.01359 | 9.83 | 1.26 | 0.049 | |||
| BD*( 1) C 5 - H 10 | 0.01155 | 9.41 | 1.19 | 0.046 | |||
| BD*( 1) C 6 - H 11 | 0.01105 | 4.77 | 1.19 | 0.033 | |||
| σ -σ* | BD ( 1) C 1 - H 7 | 1.98106 | BD*( 1) C 1 - C 2 | 0.01907 | 4.35 | 1.1 | 0.03 |
| BD*( 1) C 2 - C 3 | 0.02874 | 17.82 | 1.11 | 0.062 | |||
| BD*( 1) C 5 - C 6 | 0.01359 | 13.85 | 1.1 | 0.054 | |||
| σ -σ* | BD ( 1) C 2 - C 3 | 1.96808 | BD*( 1) C 1 - C 2 | 0.01907 | 14.94 | 1.27 | 0.06 |
| BD*( 1) C 1 - H 7 | 0.01199 | 8.24 | 1.19 | 0.043 | |||
| BD*( 1) C 3 - C 4 | 0.03301 | 19.04 | 1.29 | 0.068 | |||
| BD*( 1) C 3 - C 8 | 0.02874 | 17.49 | 1.28 | 0.065 | |||
| BD*( 1) C 4 - C 9 | 0.0215 | 10.46 | 1.28 | 0.051 | |||
| BD*( 1) C 8 - C 13 | 0.01908 | 10.71 | 1.27 | 0.051 | |||
| BD*( 1) N 17 - H 19 | 0.01237 | 4.39 | 1.18 | 0.032 | |||
| σ -σ* | BD ( 1) C 2 - N 17 | 1.98308 | BD*( 1) C 1 - C 6 | 0.01374 | 6.74 | 1.29 | 0.041 |
| BD*( 1) C 2 - C 3 | 0.02874 | 4.23 | 1.3 | 0.032 | |||
| BD*( 1) C 3 - C 4 | 0.03301 | 9.79 | 1.31 | 0.05 | |||
| BD*( 1) C 21 - C 23 | 0.03162 | 8.16 | 1.11 | 0.042 | |||
| σ -σ* | BD ( 1) C 3 - C 4 | 1.95847 | BD*( 1) C 2 - C 3 | 0.02874 | 19.54 | 1.26 | 0.069 |
| BD*( 1) C 2 - N 17 | 0.02361 | 15.23 | 1.04 | 0.055 | |||
| BD*( 1) C 3 - C 8 | 0.02874 | 19.54 | 1.26 | 0.069 | |||
| BD*( 1) C 4 - C 5 | 0.0215 | 14.56 | 1.26 | 0.06 | |||
| BD*( 1) C 4 - C 9 | 0.0215 | 14.56 | 1.26 | 0.06 | |||
| BD*( 1) C 5 - H 10 | 0.01155 | 7.87 | 1.19 | 0.043 | |||
| BD*( 1) C 8 - N 18 | 0.02365 | 15.23 | 1.04 | 0.055 | |||
| BD*( 1) C 9 - H 14 | 0.01154 | 7.87 | 1.19 | 0.043 | |||
| π -π* | BD ( 2) C 3 - C 4 | 1.5483 | BD*( 2) C 1 - C 2 | 0.3377 | 95.1 | 0.27 | 0.072 |
| BD*( 2) C 5 - C 6 | 0.29421 | 63.55 | 0.28 | 0.06 | |||
| BD*( 2) C 8 - C 13 | 0.33793 | 95.23 | 0.27 | 0.072 | |||
| BD*( 2) C 9 - C 12 | 0.29421 | 63.64 | 0.28 | 0.06 | |||
| σ -σ* | BD ( 1) C 3 - C 8 | 1.96807 | BD*( 1) C 1 - C 2 | 0.01907 | 10.71 | 1.27 | 0.051 |
| BD*( 1) C 2 - C 3 | 0.02874 | 17.45 | 1.28 | 0.065 | |||
| BD*( 1) C 3 - C 4 | 0.03301 | 19.04 | 1.29 | 0.068 | |||
| BD*( 1) C 4 - C 5 | 0.0215 | 10.46 | 1.28 | 0.051 | |||
| BD*( 1) C 8 - C 13 | 0.01908 | 14.94 | 1.27 | 0.06 | |||
| BD*( 1) C 13 - H 16 | 0.01201 | 8.24 | 1.19 | 0.043 | |||
| BD*( 1) N 18 - H 20 | 0.0124 | 4.39 | 1.18 | 0.032 | |||
| σ -σ* | BD ( 1) C 12 - C 13 | 1.97803 | BD*( 1) C 8 - C 13 | 0.01908 | 12.22 | 1.26 | 0.054 |
| BD*( 1) C 8 - N 18 | 0.02365 | 17.74 | 1.04 | 0.059 | |||
| BD*( 1) C 9 - C 12 | 0.01359 | 9.83 | 1.26 | 0.049 | |||
| BD*( 1) C 9 - H 14 | 0.01154 | 9.41 | 1.19 | 0.046 | |||
| BD*( 1) C 12 - H 15 | 0.01106 | 4.77 | 1.19 | 0.033 | |||
| BD*( 1) C 13 - H 16 | 0.01201 | 5.86 | 1.18 | 0.036 | |||
| σ -σ* | BD ( 1) C 12 - H 15 | 1.98314 | BD*( 1) C 4 - C 9 | 0.0215 | 14.48 | 1.11 | 0.055 |
| BD*( 1) C 8 - C 13 | 0.01908 | 15.36 | 1.1 | 0.057 | |||
| σ -σ* | BD ( 1) C 13 - H 16 | 1.98107 | BD*( 1) C 3 - C 8 | 0.02874 | 17.82 | 1.11 | 0.062 |
| BD*( 1) C 8 - C 13 | 0.01908 | 4.35 | 1.1 | 0.03 | |||
| BD*( 1) C 9 - C 12 | 0.01359 | 13.81 | 1.1 | 0.054 | |||
| σ -σ* | BD ( 1) N 17 - H 19 | 1.97746 | BD*( 1) C 2 - C 3 | 0.02874 | 13.47 | 1.22 | 0.056 |
| BD*( 1) N 18 - C 21 | 0.02797 | 14.23 | 0.97 | 0.051 | |||
| σ -σ* | BD ( 1) N 17 - C 21 | 1.98137 | BD*( 1) C 1 - C 2 | 0.01907 | 10.59 | 1.28 | 0.051 |
| BD*( 1) N 18 - H 20 | 0.0124 | 6.15 | 1.19 | 0.037 | |||
| BD*( 1) C 23 - C 24 | 0.02096 | 7.53 | 1.28 | 0.043 | |||
| σ -σ* | BD ( 1) N 18 - H 20 | 1.97748 | BD*( 1) C 3 - C 8 | 0.02874 | 13.43 | 1.22 | 0.056 |
| BD*( 1) N 17 - C 21 | 0.02799 | 14.18 | 0.97 | 0.051 | |||
| σ -σ* | BD ( 1) N 18 - C 21 | 1.98135 | BD*( 1) C 8 - C 13 | 0.01908 | 10.59 | 1.28 | 0.051 |
| BD*( 1) N 17 - H 19 | 0.01237 | 6.15 | 1.19 | 0.037 | |||
| BD*( 1) C 23 - C 25 | 0.02102 | 7.53 | 1.28 | 0.043 | |||
| σ -π* | BD ( 1) C 21 - H 22 | 1.98147 | BD*( 2) C 23 - C 24 | 0.3528 | 12.09 | 0.56 | 0.039 |
| σ -σ* | BD ( 1) C 21 - C 23 | 1.97233 | BD*( 1) C 2 - N 17 | 0.02361 | 8.87 | 0.98 | 0.041 |
| BD*( 1) C 8 - N 18 | 0.02365 | 8.87 | 0.98 | 0.041 | |||
| BD*( 1) C 23 - C 24 | 0.02096 | 7.07 | 1.18 | 0.04 | |||
| BD*( 1) C 23 - C 25 | 0.02102 | 7.11 | 1.18 | 0.04 | |||
| BD*( 1) C 24 - C 26 | 0.01485 | 9.25 | 1.19 | 0.046 | |||
| BD*( 1) C 25 - C 28 | 0.0129 | 9.08 | 1.18 | 0.045 | |||
| σ -σ* | BD ( 1) C 23 - C 24 | 1.97551 | BD*( 1) C 21 - C 23 | 0.03162 | 6.9 | 1.09 | 0.038 |
| BD*( 1) C 23 - C 25 | 0.02102 | 15.73 | 1.27 | 0.062 | |||
| BD*( 1) C 24 - C 26 | 0.01485 | 11.59 | 1.27 | 0.053 | |||
| BD*( 1) C 24 - H 27 | 0.01386 | 5.73 | 1.19 | 0.036 | |||
| BD*( 1) C 25 - H 29 | 0.01346 | 8.87 | 1.19 | 0.045 | |||
| BD*( 1) C 26 - H 31 | 0.01551 | 9.12 | 1.19 | 0.046 | |||
| π -σ* | BD ( 2) C 23 - C 24 | 1.6656 | BD*( 1) N 17 - C 21 | 0.02799 | 6.19 | 0.58 | 0.029 |
| BD*( 1) C 21 - H 22 | 0.04118 | 9.41 | 0.74 | 0.04 | |||
| BD*( 2) C 25 - C 28 | 0.33945 | 86.73 | 0.28 | 0.068 | |||
| BD*( 2) C 26 - C 30 | 0.38222 | 80.63 | 0.27 | 0.065 | |||
| σ -σ* | BD ( 1) C 23 - C 25 | 1.97568 | BD*( 1) C 21 - C 23 | 0.03162 | 6.9 | 1.09 | 0.038 |
| BD*( 1) C 23 - C 24 | 0.02096 | 15.77 | 1.27 | 0.062 | |||
| BD*( 1) C 24 - H 27 | 0.01386 | 8.95 | 1.19 | 0.045 | |||
| BD*( 1) C 25 - C 28 | 0.0129 | 11.05 | 1.26 | 0.052 | |||
| BD*( 1) C 25 - H 29 | 0.01346 | 5.77 | 1.19 | 0.036 | |||
| BD*( 1) C 28 - H 32 | 0.01141 | 8.49 | 1.19 | 0.044 | |||
| σ -σ* | BD ( 1) C 24 - C 26 | 1.97407 | BD*( 1) C 21 - C 23 | 0.03162 | 14.77 | 1.09 | 0.055 |
| BD*( 1) C 23 - C 24 | 0.02096 | 13.47 | 1.26 | 0.057 | |||
| BD*( 1) C 24 - H 27 | 0.01386 | 5.86 | 1.18 | 0.036 | |||
| BD*( 1) C 26 - C 30 | 0.0264 | 12.09 | 1.25 | 0.054 | |||
| BD*( 1) C 26 - H 31 | 0.01551 | 6.61 | 1.19 | 0.039 | |||
| BD*( 1) C 30 - O 33 | 0.03116 | 16.61 | 0.99 | 0.056 | |||
| σ -σ* | BD ( 1) C 24 - H 27 | 1.97987 | BD*( 1) N 18 - H 20 | 0.0124 | 4.48 | 1.02 | 0.03 |
| BD*( 1) C 23 - C 24 | 0.02096 | 5.27 | 1.11 | 0.033 | |||
| BD*( 1) C 23 - C 25 | 0.02102 | 16.95 | 1.11 | 0.06 | |||
| BD*( 1) C 26 - C 30 | 0.0264 | 14.27 | 1.09 | 0.055 | |||
| σ -σ* | BD ( 1) C 25 - C 28 | 1.97445 | BD*( 1) C 21 - C 23 | 0.03162 | 14.94 | 1.09 | 0.056 |
| BD*( 1) C 23 - C 25 | 0.02102 | 13.22 | 1.26 | 0.056 | |||
| BD*( 1) C 25 - H 29 | 0.01346 | 5.69 | 1.18 | 0.036 | |||
| BD*( 1) C 28 - C 30 | 0.02221 | 10.42 | 1.25 | 0.05 | |||
| BD*( 1) C 28 - H 32 | 0.01141 | 5.56 | 1.19 | 0.036 | |||
| BD*( 1) C 30 - O 33 | 0.03116 | 18.45 | 0.98 | 0.059 | |||
| π -π* | BD ( 2) C 25 - C 28 | 1.68745 | BD*( 2) C 23 - C 24 | 0.3528 | 83.05 | 0.28 | 0.068 |
| BD*( 2) C 26 - C 30 | 0.38222 | 88.99 | 0.27 | 0.069 | |||
| σ -σ* | BD ( 1) C 25 - H 29 | 1.97956 | BD*( 1) N 17 - H 19 | 0.01237 | 4.48 | 1.02 | 0.03 |
| BD*( 1) C 23 - C 24 | 0.02096 | 17.07 | 1.11 | 0.06 | |||
| BD*( 1) C 23 - C 25 | 0.02102 | 5.27 | 1.11 | 0.033 | |||
| BD*( 1) C 28 - C 30 | 0.02221 | 14.1 | 1.1 | 0.054 | |||
| σ -σ* | BD ( 1) C 26 - C 30 | 1.98073 | BD*( 1) C 24 - C 26 | 0.01485 | 11.51 | 1.28 | 0.053 |
| BD*( 1) C 24 - H 27 | 0.01386 | 9.12 | 1.2 | 0.046 | |||
| BD*( 1) C 26 - H 31 | 0.01551 | 6.23 | 1.2 | 0.038 | |||
| BD*( 1) C 28 - C 30 | 0.02221 | 14.23 | 1.27 | 0.059 | |||
| BD*( 1) C 28 - H 32 | 0.01141 | 8.54 | 1.21 | 0.044 | |||
| π -π* | BD ( 2) C 26 - C 30 | 1.6575 | BD*( 2) C 23 - C 24 | 0.3528 | 84.22 | 0.29 | 0.069 |
| BD*( 2) C 25 - C 28 | 0.33945 | 78.91 | 0.29 | 0.066 | |||
| σ -σ* | BD ( 1) C 26 - H 31 | 1.97659 | BD*( 1) C 23 - C 24 | 0.02096 | 14.43 | 1.11 | 0.055 |
| BD*( 1) C 24 - C 26 | 0.01485 | 4.23 | 1.11 | 0.03 | |||
| BD*( 1) C 26 - C 30 | 0.0264 | 4.48 | 1.09 | 0.031 | |||
| BD*( 1) C 28 - C 30 | 0.02221 | 15.86 | 1.1 | 0.058 | |||
| BD*( 1) C 34 - H 35 | 0.01969 | 4.52 | 1.01 | 0.03 | |||
| σ -σ* | BD ( 1) C 28 - C 30 | 1.97936 | BD*( 1) C 25 - C 28 | 0.0129 | 9.75 | 1.28 | 0.049 |
| BD*( 1) C 25 - H 29 | 0.01346 | 9.12 | 1.2 | 0.046 | |||
| BD*( 1) C 26 - C 30 | 0.0264 | 13.97 | 1.26 | 0.058 | |||
| BD*( 1) C 26 - H 31 | 0.01551 | 8.87 | 1.2 | 0.045 | |||
| BD*( 1) C 28 - H 32 | 0.01141 | 5.86 | 1.21 | 0.037 | |||
| BD*( 1) O 33 - C 34 | 0.00541 | 6.11 | 0.99 | 0.034 | |||
| σ -σ* | BD ( 1) C 28 - H 32 | 1.97995 | BD*( 1) C 23 - C 25 | 0.02102 | 15.27 | 1.1 | 0.057 |
| BD*( 1) C 26 - C 30 | 0.0264 | 16.9 | 1.08 | 0.059 | |||
| σ -σ* | BD ( 1) C 30 - O 33 | 1.98907 | BD*( 1) C 24 - C 26 | 0.01485 | 5.94 | 1.39 | 0.04 |
| BD*( 1) C 25 - C 28 | 0.0129 | 5.86 | 1.38 | 0.039 | |||
| BD*( 1) C 34 - H 36 | 0.00893 | 5.94 | 1.3 | 0.038 | |||
| σ -σ* | BD ( 1) O 33 - C 34 | 1.99038 | BD*( 1) C 28 - C 30 | 0.02221 | 11.63 | 1.35 | 0.055 |
| σ -σ* | BD ( 1) C 34 - H 35 | 1.99375 | BD*( 1) C 26 - H 31 | 0.01551 | 6.02 | 1.04 | 0.035 |
| σ -σ* | BD ( 1) C 34 - H 36 | 1.99106 | BD*( 1) C 30 - O 33 | 0.03116 | 11.55 | 0.83 | 0.043 |
| n -π* | LP ( 1) N 17 | 1.86213 | BD*( 2) C 1 - C 2 | 0.3377 | 70.21 | 0.35 | 0.072 |
| BD*( 1) N 18 - C 21 | 0.02797 | 7.24 | 0.65 | 0.031 | |||
| BD*( 1) C 21 - H 22 | 0.04118 | 24.81 | 0.81 | 0.063 | |||
| n -π* | LP ( 1) N 18 | 1.86229 | BD*( 2) C 8 - C 13 | 0.33793 | 70.17 | 0.35 | 0.072 |
| BD*( 1) N 17 - C 21 | 0.02799 | 7.24 | 0.65 | 0.031 | |||
| BD*( 1) C 21 - H 22 | 0.04118 | 24.81 | 0.81 | 0.063 | |||
| n -σ* | LP ( 1) O 33 | 1.96907 | BD*( 1) C 26 - C 30 | 0.0264 | 21.13 | 1.13 | 0.068 |
| BD*( 1) C 34 - H 35 | 0.01969 | 7.2 | 1.05 | 0.038 | |||
| BD*( 1) C 34 - H 36 | 0.00893 | 4.98 | 1.07 | 0.032 | |||
| n -π* | LP ( 2) O 33 | 1.87925 | BD*( 2) C 26 - C 30 | 0.38222 | 77.86 | 0.33 | 0.075 |
| BD*( 1) C 28 - C 30 | 0.02221 | 5.36 | 0.87 | 0.031 | |||
| BD*( 1) C 34 - H 35 | 0.01969 | 16.36 | 0.78 | 0.051 | |||
| BD*( 1) C 34 - H 37 | 0.01677 | 23.97 | 0.79 | 0.062 | |||
| π*-π* | BD*( 2) C 1 - C 2 | 0.3377 | BD*( 2) C 3 - C 4 | 0.5074 | 1094.2 | 0.01 | 0.083 |
| π*-π* | BD*( 2) C 8 - C 13 | 0.33793 | BD*( 2) C 3 - C 4 | 0.5074 | 1081 | 0.01 | 0.083 |
| π*-σ* | BD*( 2) C 23 - C 24 | 0.3528 | BD*( 1) C 21 - H 22 | 0.04118 | 4.9 | 0.46 | 0.047 |
| π*-π* | BD*( 2) C 26 - C 30 | 0.38222 | BD*( 2) C 23 - C 24 | 0.3528 | 1114.1 | 0.01 | 0.079 |
In perimidine moiety, the orbitals interaction between π(C1-C2)→π* (C5-C6), π(C5-C6)→ π*(C3-C4), π(C8-C13)→π*(C9-C12), π(C9-C12)→π*(C3-C4), which transfer higher energy: 88.99, 80.63, 88.95, 80.54 KJ/mol, respectively and hence stabilization increases. It is noted from the Table--- that there occur a strong intra-molecular hyperconjugative interactions of C1-C2 and C13-C18 from π(C3-C4) → π*(C1-C2) / (C8-C13), which increase electron density (ED) 0.3377 and 0.3379 e) that weakens the respective bands C1-C2 and C8-C13 leading to the stabilization of 95.1 and 95.23KJ/mol, respectively. The anti-bonding orbitals (C1-C2) and (C13-C8) are attached with N17 and N18 atoms, respectively, which also enhances the EDs due to resonance structure of the compound.
Similarly in benzene ring, the strong intra-molecular hyperconjugative interactions: π(C23-C24)→π*(C25-C28), π(C25-C28)→π*(C26-C30), π(C26-C30)→π*(C23-C24), which increase the electron densities (EDs): (0.3395, 0.3822, 0.3528e), that weaken the respective bonds and hence the stabilization increases 86.73, 88. 99, 84.22 KJ/mol, respectively.
The lone pair of oxygen and nitrogen atoms play great role in the MPDP molecule. During n→π* transitions:LpN17→π* (C1-C2) and LpN18→π* (C8-C13) more energy delocalization take place and their corresponding excitation energy values are: 70.21 and 70.17KJ/mol, respectively. Besides the Lp O33 atom transfer energy: 77.86KJ/mol to anti-bonding orbital π* (C26-C30) on comparing with π* (C28-C30) transition: 5.36KJ/mol. This is due to the lone pair electrons of oxygen in methoxy group may be involved in energy state. So that abnormal energy is appeared. The maximum hyperconjugative E(2) energy of heteroatoms during the intra-molecular interaction leads the molecule towards medicinal and biological applications.
Homo-Lumo Analysis
The frontier molecular orbitals play an important role in the optical and electric properties, as well as in quantum chemistry. The frontier molecular orbital gap also helps to characterize the chemical reactivity and the kinetic stability of the molecule. A molecule with a small frontier orbital gap is generally associated with high chemical reactivity and low kinetic stability and is also termed as soft molecule 51. The calculated energy values of HOMO and LUMO in gas phase are -5.0725 and -1.0149eV, respectively, and the frontier orbital energy gap value is 3.0437eV. The frontier molecular orbitals (HOMO and LUMO) and their corresponding energies are shown in Figure 5. physico chemical parameters are shown in Table 6. The density of state (DOS) of the present molecule has been plotted and shown in Figure 6. DOS denoted the number of available molecular orbitals at different energy.
Table 6. The Physico-chemical properties of MPDP| Parameters | Values |
| HOMO | -5.072 eV |
| LUMO | -1.014 eV |
| Energy gap | 3.043 eV |
| Ionization potential (IP) | -5.072 eV |
| Electron affinity (EA) | -1.014 eV |
| Electrophilicity Index (ω) | 1.141 |
| Chemical Potential (µ) | 3.043 |
| Electronegativity (χ) | -3.043eV |
| Hardness (η) | -3.043 |
Figure 5.The frontier molecular orbital diagram of MPDP
Figure 6.The Dos spectrum
UV–Vis Analysis
The absorption wavelengths (λ), oscillator strengths (f), and excitation energies were calculated at TD-DFT/B3LYP/6-311++G(d,p) level. The UV–Vis spectrum of the MPDP was recorded in the wavelength range 200–800nm as shown in Figure 7. The electronic spectrum of title molecule shows the transitions at 276nm and 327nm. In MPDP, the calculated UV absorption result has three excited states (ESs). The ES1, ES2, and ES3 appeared at 342.36, 313.62 and 309.46 nm and their corresponding excitation energies are 3.6214, 3.9533 and 4.006eV, respectively. These bonds are due to transition HOMO→LUMO (88%), HOMO→LUMO-3(80%) and HOMO→LUMO-2(92%) in gaseous phase. The calculated band gap 309.46 nm of ES3 coincide with the recorded value of 276 nm. The excitation energies of MPDP are listed in Table 7.
Figure 7.Combined experimental and theoretical UV-Visible spectra of MPDP
| Calculated at B3LYP/6-311++G(d,p) | Oscillator strength | Calculated Band gap(eV/nm) | Experimental Band gap(eV/nm) |
| Excited State 1 73 -> 74 | Singlet-A(f=0.1842) 0.66291 | 3.6214 eV/342.36 nm 4.057476 | 327 |
| Excited State 2 70 -> 74 70 -> 75 73 -> 76 | Singlet-A(f=0.0611) -0.24099 0.12032 0.63209 | 3.9533 eV/313.62 nm -5.644712 6.062133 4.611499 | |
| Excited State 3 3 -> 75 73 -> 77 | Singlet-A(f=0.0160) 0.67644 -0.10991 | 4.0065 eV/309.46 nm 4.474898 -4.661295 | 276 |
Mulliken Charges
The calculation of atomic charges plays an important role in the application of quantum mechanical calculations to molecular systems 52. The charge distributions are calculated by Mulliken method using B3LYP/6-311++G(d,p) level of basis set for the equilibrium geometry of DPMP are given in Table 8. The charge distribution on the molecule has an important influence on the vibrational spectra. The corresponding Mulliken’s plot is shown in Figure 8. The C4 atom possess most positive charge, which is due to the formation of conjugation in perimidine ring. The C21 atom attached with two nitrogen atom in adjacent position. Due to the presence of lone pair of electrons on nitrogen atoms, the C21 atom has more negative charge among the other carbon atoms. All the hydrogen atoms have net positive charges.
Table 8. The Mulliken atomic charges of MPDP| Atoms | Charges |
| 1C | -0.1527 |
| 2C | -0.2458 |
| 3C | 0.3349 |
| 4C | 0.4394 |
| 5C | -0.3593 |
| 6C | -0.1967 |
| 7H | 0.1148 |
| 8C | -0.2458 |
| 9C | -0.3593 |
| 10H | 0.1225 |
| 11H | 0.1660 |
| 12C | -0.1968 |
| 13C | -0.1529 |
| 14H | 0.1225 |
| 15H | 0.1660 |
| 16H | 0.1148 |
| 17N | 0.2352 |
| 18N | 0.2352 |
| 19H | 0.2302 |
| 20H | 0.2301 |
| 21C | -0.8104 |
| 22H | 0.1968 |
| 23C | 0.0076 |
| 24C | 0.3594 |
| 25C | -0.1828 |
| 26C | 0.1642 |
| 27H | 0.1921 |
| 28C | -0.3033 |
| 29H | 0.1628 |
| 30C | -0.7835 |
| 31H | 0.2038 |
| 32H | 0.2025 |
| 33O | -0.1541 |
| 34C | -0.3411 |
| 35H | 0.1531 |
| 36H | 0.1775 |
| 37H | 0.1531 |
Figure 8.Mulliken atomic charges plot of MPDP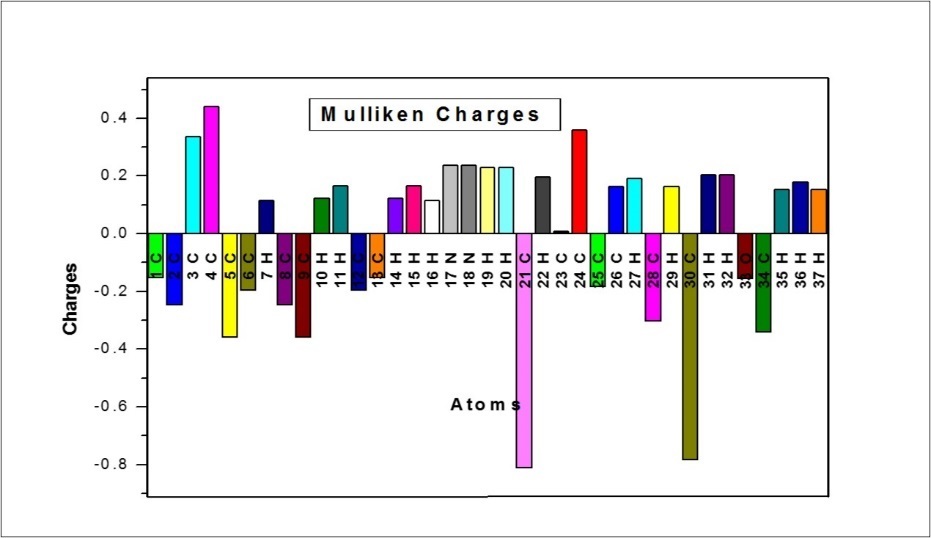
MEP Analysis
In the present study, 3D plots of molecular electrostatic potential (MEP) has been drawn in Figure 9. The MEP is a plot of electrostatic potential mapped onto the constant ED surface. The different values of the electrostatic potential at the surface are represented by different colors. Potential increases in the order red < orange < yellow < green < blue. The color code of the MEP map is in the range between -9.517e-2 (deepest red) and 9.517e-2 (deepest blue) and the, blue colour represents the strongest attraction and red represents the strongest repulsion. The MEP map shows, the negative region is located on nitrogen atom in perimidine ring (red region) and the positive region (blue region) is located on the methoxy group is phenyl ring.
Figure 9.MEP surfaces map of MPDP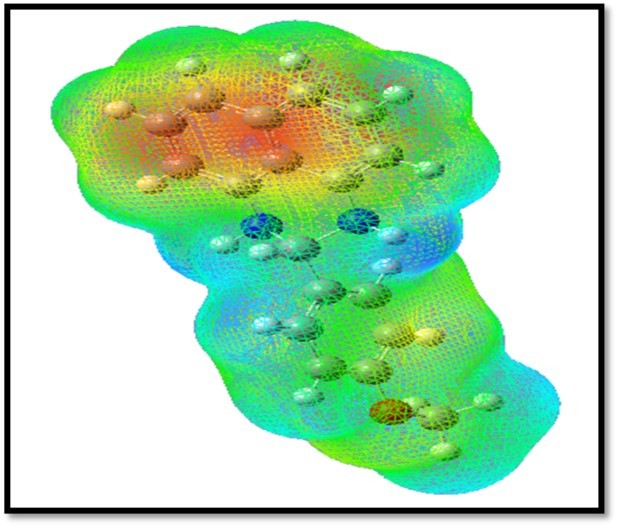
The red and blue areas in the MEP map refer to the regions of negative and positive potentials and correspond to the electron rich and electron-poor regions, respectively, whereas the green color denotes the neutral electrostatic potential. The MEP surface provides necessary information about the reactive sites.
Molecular Docking:
Molecular docking study was carried out to study the precise binding site of ligand on protein. A molecular docking study is a key tool in structural molecular biology and computer assisted drug design. The synthesized analogue docking with crystal structure of human lanosterol 14-alpha dimethylase in complex with ketaconazole (3LD6). Analogue showed best ligand pose energy -13.23kcal/mol in 3LD6 protein. The protein ligand interactions are shown in Figure 10. In compound MPDP surrounded by wander walls interaction of amino acids residues PHEB543, METB545, ALAB682, HISB681 and METB932. There are two alkyl interaction TRPB684 and LEUB685 inside the benzene ring. The pi-alkyl interaction in PHEB550, METB826, PHEB552, CYSB847, TYRB552 and ILEB824 perimidine inside the ring. From this interaction it can be predicted as the activity may be due to inhibition of human lanosterol 14-alpha dimethylase in complex with ketaconazole.
Figure 10.2d and 3d interaction of compound MPDP
Thermodynamic Properties
The thermodynamic functions such as entropy (S), heat capacity at constant pressure (Cp) total and enthalpy (E) for different range (100–1000 K) of temperatures are determined and these results are presented in the Table 9. The thermodynamic functions are increasing with temperature ranging from 100 to 1000 K due to the fact that the molecular vibrational intensities increase with temperature 53. The correlation graph between thermodynamic functions and its different temperatures are graphically represented in Figure 11. All the thermodynamic data supply helpful information for the further study on the molecule MPDP. They can be used to compute the other thermodynamic energies according to relationships of thermodynamic functions and estimate directions of chemical reactions according to the second law of thermodynamics in thermochemical field.
Table 9. Thermodynamic properties at different temperatures of MPDP| T (K) | S (J/mol.K) | Cp (J/mol.K) | ddH (kJ/mol) |
| 100 | 355.52 | 110.89 | 7.13 |
| 200 | 459.34 | 202.39 | 22.63 |
| 298.15 | 558.88 | 303.23 | 47.43 |
| 300 | 560.76 | 305.12 | 47.99 |
| 400 | 662.01 | 401.14 | 83.42 |
| 500 | 760.43 | 481.08 | 127.67 |
| 600 | 854.01 | 544.97 | 179.1 |
| 700 | 942 | 596.07 | 236.24 |
| 800 | 1024.39 | 637.57 | 297.99 |
| 900 | 1101.53 | 671.79 | 363.52 |
| 1000 | 1173.83 | 700.37 | 432.17 |
The corresponding fitting equations are as follows:
C0p,m = 7.1574 + 0.0302T + 2.6697x10-5 T2 (R2 = 0.9992)
S0m = 3.3949 + 0.0143T + 1.2663x10-5 T2 (R2 = 0.9999)
ΔH0m = 4.8494 + 0.0204T + 1.8089x10-5 T2 (R2 = 0.9993)
All the given thermodynamic data are the helpful information for further study on MPDP. It can be used to compute the other thermodynamic energies according to relationships of thermodynamic functions and estimate directions of chemical reactions according to the second law of thermodynamics in thermochemical field 53. All the thermodynamic calculations were done in gas phase and they could not be used in solution.
Figure 11.Correlation graphs between thermodynamic functions VS temperatures of MPDP
Conclusion
MPDP Compound was synthesized and characterized by FT-IR, FT-Raman, UV-Vis, and NMR spectra. The calculated geometrical parameters and harmonic frequencies were in good agreement with literature values. A complete vibrational analysis was made for the first time to the molecule MPDP. The N17-C21-N18 ring portion attained twisted boat configuration due to steric effect. The first order hyperpolarizability (β0) value is eighteen times greater than that of urea and hence the present molecule possesses good NLO property. The LP O33 atom transfer more energy: 77.86 kj/mol to anti-bonding orbital π* (C26-C30), which may be due to the involvement of lone pair electrons in oxygen of methoxy group in energy state. The calculated band gap energy was 4.057ev. The calculated UV-Vis spectral values are in good agreement with the observed values. The absorption maximum 276 was assigned to π-π* type. The MEP surface plot represents the reactive sites of nucleophilic and electrophilic attack. The thermodynamic parameters (entropy, enthalpy and heat capacity) are calculated in the temperature range from 100 to 1000k. In addition Mulliken atomic charges and various physico-chemical properties are also calculated. The docking study indicate that the molecule MPDP possessis high binding energy -13.23kcal/mol, which may be due to the Vander walls and π-alkyl interactions.
References
- 1.J S Casas, Castellano E E, M D Couce, Ellena J, Sánchez A et al. (2006) A gold(I) complex with a vitamin K3 derivative: Characterization and antitumoral activity. , J. Ing. Bio.Chem 100, 1858-1860.
- 2.D X West, A E Liberta, S B Padhye, R C Chikate, P B Sonawane et al. (1993) Thiosemicarbazone complexes of copper(II): structural and biological studies. , Coord. Chem. Reviews 123, 49-71.
- 3.M C Rodriguez-Argüelles, M B Ferrari, C Pelizzi Fava, Tarasconi G G, Albertini P et al. (1995) 2,6-Diacetylpyridine bis(thiosemicarbazones) zinc complexes: Synthesis, structure, and biological activity. , J. Inorg. Biochem 58, 157-175.
- 4.Casas J, M S García-Tasende, Maichle-Mössmer C, M C Rodríguez-Argüelles, Sánchez A et al. (1996) synthesis, structure, and spectroscopic properties of acetato (dimethyl) (pyridine-2-carbaldehydethiosemicarbazonato)tin(IV) acetic acid solvate, [SnMe2 (PyTSC)(OAc)].HOAc. Comparison of its biological activity with that of some structurally related diorganotin (IV) bis (thiosemicarbazonates). , J. Inorg. Biochem 62, 41-55.
- 5.M B Ferrari, Fava G G, Tarasconi P, Albertini R, Pinelli S et al. (1994) Synthesis, spectroscopic and structural characterization, and biological activity of aquachloro (pyridoxal thiosemicarbazone) copper(II) chloride. , J. Inorg. Biochem 53, 13-25.
- 6.Koch U, Attenni B, Malancona S, Colarusso S, Conte I et al. (2006) 2-(2-Thienyl)-5,6-dihydroxy-4-carboxypyrimidines as Inhibitors of the Hepatitis C Virus NS5B Polymerase: Discovery,SAR,Modeling, and Mutagenesis. , J. Med. Chem 49, 1693-1705.
- 7.Zamora F, Kunsman M, Sabat M, Lippert B. (1997) Metal-Stabilized Rare Tautomers of Nucleobases. 6. † Imino Tautomer of Adenine in a Mixed-Nucleobase Complex of Mercury (II). , Inorg. Chem 36, 1583-1587.
- 8.Jolibois F, Cadet J, Grand A, Subra R, Rega N et al. (1998) Structures and Spectroscopic Characteristics of 5,6-Dihydro-6-thymyl and 5,6-Dihydro-5-thymyl Radicals by an Integrated Quantum Mechanical Approach Including Electronic, Vibrational, and Solvent Effects. , J. Amer. Chem. Soc 120, 1864-1871.
- 10.Pokladko M, Gondek E, Sanetra J, Nizioł J, Danel A et al. (2009) Spectral emission properties of 4-aryloxy-3-methyl-1-phenyl-1H-pyrazolo [3, 4-b] quinolines, Spectrochim. Acta Part A: Molecularand Bio.Spectro. 73, 281-285.
- 11.Fuks-Janczarek I, A H Reshak, Kuźnik W, I V Kityk, Gabański R et al. (2009) UV–vis absorption spectra of 1,,-dialkoxy-2,5-bis[2-(thien-2-yl)ethenyl]benzenes. , Spectrochim. Acta Part A: Mol. Biomol. Spectros 72, 394-398.
- 12.A H Reshak, Stys D, Auluck S, I V Kityk. (2010) Linear and Nonlinear Optical Susceptibilities of 3-Phenylamino-4-phenyl-1, 2. , 4-triazole-5-thione, J. Phys. Chem 114, 1815-1821.
- 13.A H Reshak, Auluck S, Stys D, I V Kityk, Kamarudin H et al. (2011) Dispersion of linear and non-linear optical susceptibilities for amino acid 2-aminopropanoic CH3CH(NH2)COOH single crystals: experimental and theoretical investigations,”. , J. Mater. Chem 21, 17219.
- 14.A H Reshak, Kamarudin H, Auluck S. (2012) Acentric Nonlinear Optical 2,4-Dihydroxyl Hydrazone Isomorphic Crystals with Large Linear, Nonlinear Optical Susceptibilities and Hyperpolarizability. , J. Phys. Chem 116, 4677-4683.
- 15.A H Reshak, Kamarudin H, Auluck S. (2013) Electronic structure, density of electronic states, and the chemical bonding properties of 2,4-dihydroxyl hydrazone crystals (C13H11N3O4). , J. Mater. Sci 48, 3805-3811.
- 16.A H Reshak, Kamarudin H, I V Kityk, Auluck S. (2013) Electronic structure, charge density, and chemical bonding properties of C11H8N2O o-methoxydicyanovinylbenzene (DIVA) single crystal. , J.ournal of Materials Science 48, 5157-5162.
- 17.V M Chernyshev, D A Pyatakov, A N Sokolov, Astakhov3 A V, Gladkov E S et al. (2014) Partially hydrogenated 2-amino[1,2,4]triazolo[1,5-a]pyrimidines a synthons for the preparation of polycondensed heterocycles: reaction with chlorocarboxylic acid chlorides. , Tetrahedron 70, 684-701.
- 18.Kanbara T, Kushida T, Saito N, Kuwajima I, Kubota K et al. (1992) . Preparation and Properties of Highly Electron-accepting Poly (pyrimidine-2,5-diyl,” Chem. Lett 21, 583-586.
- 19.Meyer T.J.(1989May)Chemical approaches to artificial photosynthesis,”. , Acc. Chem. Res 22, 163-170.
- 20.A I Pavluchenko, V F Petrov, N I Smirnova. (1995) Liquid crystalline 2,5-disubstituted pyridine derivatives,” Liquid Crystals. 19, 811-821.
- 22.Asghari S, Tajbakhsh M, B J Kenari, Khaksar S. (2011) Supramolecular synthesis of 3, 4-dihydropyrimidine-2(1H)-one/thiones under neat conditions. , Chinese Chem. Lett 22, 127-130.
- 23.Alam M, D U Lee. (2016) synthesis, spectroscopic and computational studies of 2-thiophen-2yl)-2, 3-dihydro-1H-Perimidine: An enzymes inhibition study,” computational biology and chemistry. 64, 185-201.
- 24.Wasulko W, A C Noble, F D Popp. (1966) Synthesis of potential antineoplastic agents XIV. Some 2-substituted 2, 3-dihydro-1H-perimidines. , J. Med. Chem 9, 599-601.
- 26.H B Schlegel. (1982) Optimization of equilibrium geometries and transition structures,”. , J. Com. Chem 3, 214-218.
- 30.Michalska D, Wysokinski R. (2005) The prediction of Raman spectra of platinum (II) anticancer drugs by density functional theory,”. , Chem. Phys. Lett 403, 211-217.
- 32.Rauhut G, Pulay P. (1995) transferable Scaling Factors for Density Functional Derived Vibrational Force Fields,”. , J. Phys. Chem 99, 3093-3100.
- 33.Li Z, Deng W. (2011) Synthesis, characterization, crystal structure and DFT studies on 1′, 3′-dihydrospiro [fluorene-9, 2′-perimidine,” Spectrochim. Acta Part A:. , Mol. Biomol. Spectros 82, 56-62.
- 34.Socrates G. (1980) Infrared characteristic group Frequencies 3rd Ed., Wiley Interscience,” Pubs. , New York
- 35.Socrates G. (2001) Infrared and Raman characteristic group frequencies-Tables and charts 3rd Ed. , Wiley: New York
- 36.Meganathan C, Sebastian S, Kurt M, Lee Woo, Sundaraganesan K et al. (2010) Molecular structure, spectroscopic (FTIR, FTIR gas phase, FT-Raman) first-order hyperpolarizability and HOMO-LUMO analysis of 4-methoxy-2-methyl benzoic acid,”. , J. Raman Spectrosc 41, 1369-1378.
- 37.Lakshmaiah B, Rao Ramana, G. (1989) Vibrational analysis of substituted anisoles. I-Vibrational spectra and normal coordinate analysis of some fluoro and chloro compounds,”. , J. Raman Spectrosc 20, 439-448.
- 39.B Venkataram Reddy, Rao Ramana, G. (1994) Transferable valence force fields for substituted benzenes,”. , Vibrational Spectrosc 6, 231-250.
- 40.V Ashok Babu, Lakshmaiah B, K Sree Ramulu, G Ramana Rao. (1987) . , Indian J. Pure Appl. Phys 25, 58.
- 41.Silverstein M, C G Basseler, Morill C. (1981) Spectrometric Identification of Organic Compounds,Wiley,NewYork.
- 42.A D Khalaji, A N Chermahini, Fejfarova K, Dusek M.Synthesis, characterization, crystal structure, and theoretical studies on Schiff-base compound 6-[(5-Bromopyridin-2-yl) iminomethyl] phenol. , Struct. Chem 21, 153-157.
- 43.H T Varghese, C Y Panicker, V S Madhavan, Mathew S, Vinsova J. (2009) FT-IR, FT-Raman and DFT calculations of the salicylanilide derivate 4-chloro-2-(4-bromophenylcarbamoyl phenyl acetate,”. , J. Raman Spectrosc 40, 1211-1223.
- 44.Varsanyi G. (1974) Assignments of vibrational spectra of 700 Benzene. , Derivatives,Wiley,New York
- 45.B J Coe, J A Harris, L A Jones, Brunschwig B S, Song K et al. (2005) Syntheses and Properties of Two-Dimensional Charged Nonlinear Optical Chromophores Incorporating Redox-Switchable cis -Tetraammineruthenium(II) Centers . , J. Ameri. Chem. Society 127, 4845-4859.
- 47.A E Reed, R B Weinstock, Weinhold F. (1985) Natural population analysisa. , J. Chem. Phy 83, 735-746.
- 48.A E Reed, Weinhold F. (1983) Natural bond orbital analysis of near‐Hartree–Fock water dimer,”. , J. Chem. Phy 78, 4066-4073.
- 49.J P Foster, Wienhold F. (1980) Natural hybrid orbitals. , J. Ameri. Chem. Society 102, 7211-7218.
- 50.Chocholousova J, Vladimir Spirko V, Hobza P. (2004) First local minimum of the formic acid dimer exhibits simultaneously red-shifted O–H⋯O and improper blue-shifted C–H⋯O hydrogen bonds,”. , Journal of Chemical Physics 6, 37-41.
Cited by (1)
- 1.Thirughanasambantham N., Balachandran V., Narayana B., Sivakumar C., Jayashree A., et al, 2024, Pharmaceutical investigation of 2-Amino-4-(2-fluorophenyl)-5,10-dioxo-5,10-dihydro-4H-benzo[g]chromene-3-carbonitrile via computational studies, Inorganic Chemistry Communications, 159(), 111709, 10.1016/j.inoche.2023.111709