
Human Proteome Project and Current Bioinformatics Status in Disease Diagnosis and Treatment
Abstract
Human proteome project was revolutionized about 40 years ago with purpose of summarizing whole proteomic data at one place. It was launched after human genome project to map and observe all proteins. The goal related proteomic study is to draft the entire human proteome in disease diagnosis by using bioinformatics tools. Pillars of human proteome project provide different databases related to proteins at transcriptional and translational level. Human proteome organization(HUPO) published biology disease HUPO whose aim is to measure protein and proteome by life and processes related to human diseases. Different human organ like plasma, liver, brain and diabetic base project are used to characterize human disease and health. Major data resources accumulated in databases like peptides Atlas, GPMDB and neXtProt for proteins. Matrices of human proteome project identify and characterize the protein products as Post translational modification (PTM), splice various isoforms from 20,300 proteins. Matrices related to different years make proteomes counterpart by magnify the research biomedical community with high output of instruments and specimen pre-analytical protocols. CALIPHO multidisciplinary group provides information about protein complexities, interactions, function and structure complexities after Uniport and Swissprot. Different bioinformatics tools are used for structural and functional annotations of protein, disease diagnosis and mutations due to protein. Extensive study of human proteome project has been proved helpful in disease treatment at translational and post- translational levels. In future, human proteome project along with bioinformatics will include protein profiling, biomarkers, Mass spectrophotometer technique and cross analysis of different proteome projects.
Author Contributions
Academic Editor: Bobbie-Jo M, Webb-Robertson, Senior Research Scientist Pacific Northwest National Laboratory Computational Biology and Bioinformatics Richland, WA, USA
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2018 Pervez Anwar
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
A large amount of data means that many problems faced in biology are now being faced in computing too. Bioinformatics, is the field that includes different techniques and softwares to examine and elucidate biological data. Two important wide-ranging activities that utilize bioinformatics are genomics and proteomics and help bioinformatics tools to predict about genes and proteins. The international HUPO was designed five years ago to characterize and evaluate so-called "missing proteins" those were confidently estimated but have not been detected at protein level yet. Currently, there are 2,563 such "missing proteins". To see the detailed distribution across chromosomes and protein existence status, HPP took help of CALIPHO; i.e. neXtProt to identify these proteins 1, 2, 3 .
To overcome the drawbacks of UniProt/Swiss-Prot group in 2008, a team was established known as CALIPHO. To measure these approaches, CALIPHO developed neXtProt. All the data related to human proteome is present in the data base that is neXtProt. A common development of the SIB and of Gene Bio SA. It works like the model organism database which is used for collection of data related to species and act as one of the databases which provide stimulus/input for research on model species. As like that neXtProt provide all the data for the protein present in human, to combine data correctly and develop tools that are required by the users and also provide user with high quality data and tools4, 5. Bioinformatics’ tools are used for proteome analysis to know about structure of protein, about functional annotation and to diagnose diseases and mutation due to protein. Different software’s along with their input and output details are described in this article such as PSIPRED and I- TASSER used for protein structure and COAFACTOR and JAFA used for protein functional annotation. Like this, Predict AD and BLAT used for protein disease and mutation annotation 6, 7, 8, 9. Different biomarkers present in the patient serum samples can be recorded by performing imaging test. These biomarkers help in the diagnosis of health and disease characteristics. And the next step is protein profiling that find out the comparison protein profiles with controlled alternative protein by the use of DIGE 10, 11.
HUPO with collaboration of different workgroups is making new projects to identify diseases related to human health and their identification. Approximately ~25% proteins structures, functions, localization and post translational modifications are not analyzed. So, with the passage of time new databases and softwares are developing to overcome the issue 12.
The Human Genome Project (HGP)
This is a scientific based research project aimed at, drafting the whole genome of human, and characterize the structures and functions. The Human Genome Project (HGP) idea was given in 1984 by Renato Dulbecco but the work on project was started in 1990 and after a long time of 13 years, it was completed in 2003. The main funding for the project was from US government through the National institutes of health as well as from other institutions from all over the world 13.The HGP and resulting study related to our genome has evolved the application of medicine, motivating wide range data acquisition schemes such as the 1000 Genomes Project, the Chimpanzee Genome Project, Neanderthal Genome Project, and Cancer Genome Atlas. Started in 2008 this is an international research scheme that develop whole study and record gene variation accrue in the (www.1000genomes.org). progress of HGP impressed many other researcher and create other projects such as, human proteome and brain 14.
Human Proteome Project (HPP)
After innovation of HGP, the “HUPO” propel a new project that is HPP. Its ambition is to plan and perceive all proteins, translated from sequences within human organization by using three running pillars that is MS (MASS SPECTROMETER), antibody capture and bioinformatics tools as well as knowledge bases. These pillars form footing upon which chromosomes-based HPP and the biology of diseased HPP are formulated. The C-HPP project was emanated by collaboration of five Asian countries: Thailand, Singapore, Taiwan, Hong Kong and India. Its sight is to examine all misplaced and familiar proteins which are translated by chromosome 12 for their sub-cellular localization 15, 16. The C-HPP ch12 consortium work with other C-HPP teams and existing initiatives under B/D HPP. For example Human Brain Proteome Project, Membrane Proteomic initiatives, and so on.. B/D-HPP was developed by HUPO whose intent is to support the measurement of the proteins & proteome by life and process measurement or disease related to humans.17, 18
Human Proteome Project Initiative Hupo-Psi:
Due to elevation in proteomics data, there is obligation to collect, store and manage data to make it congenial for scientist. Orders to depository have been in easy manners; it have to be represented in a particular format. The Human Proteome Organization “Proteomics Standard Initiatives” is furnishing such standards, implicating the instrumentations and development of different tools to make it easily available19. Internal structure of PSI contains certain groups (Figure 1) that generate different products based on different workshops. These instruments are particularly complex. Molecular Interactions (MI) address description of protein-protein interaction and broadened scope to encircle all types of r interactions on the level of molecular. Protein Modification workgroup propound specific study of structures and specific naming vocabulary for naratting the naturally and artificial protein modification. Single experiment cannot endow all necessary data about modifications and usually ambiguity fall in each reported modification. 20, 21
Figure 1.structure of HUPO-PSI work groups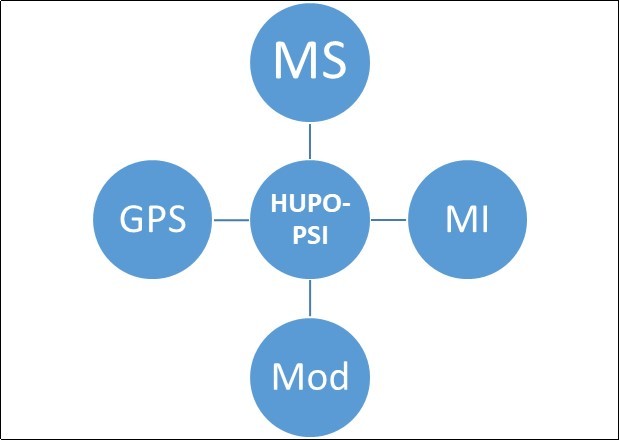
4th HUPO Diabetes Workshop, Yokohama:
The 12th HUPO that is Human Proteome Organization yearly event hosted by Yokohama (Japan). HUPO conference permits researchers initiative to share their capabilities. 90 participants were attracted towards the 4th workshop of Human Diabetes Proteome Project at HUPO 2013. 22
Human Plasma Proteome Project (HPPP):
PPP pilot phase called the “Exploring the human plasma proteome”. PPP generate a data set for 3020 proteins which point out as more than two peptides and are totally approachable at EBI/PRIDE, ISB/peptide Atlas.23. Hppp started in 2002 and in 2003 to 2005, HPPP formulate and disperse specimen of Human serum to 55 attendees research labs worldwide24, 25, 26, 27.Goals of HPPP are to analyze constituents of human plasma and serum to identify variations, causes and treatment 23. PPD gives qualitative and quantitative information about proteins which serve as reference platform for biomarkers discovery 28.
Human Liver Proteome Project (HLPP):
The liver project was the first start of HPP for organ and tissue.29 HLPP started in 2002 30. It is divided into two phases: Pilot phase aims are to arrange globally work to evaluate & construct technology platform, to produce the infra-structure for complete profiling of HLPP.29.CHNLPP was launched in November 2004 with collaboration of 50 institutions and 70 laboratories for this purpose.31, 32.Next phases of these projects are to perform functional studies and further understanding of liver biology 33.
HUPO Brain Proteome Project (BPP):
The HUPO BPP is chaired, structured and organized by Helmut E Meyer and Joachim Klose. This project started in 2002 across the world. HUPO BPP unites some research labs offer connection and interactions with newly developed neurological field. HUPO BPP is to understand the process of brain proteins in the nervous system related disease and aging. 34 Therefore, study of body serums/fluids is related to HUPO BPP 35.
In 2015, large scale and targeted state study of proteome related to human brain and body fluids occured.
The 2009 study aim was to obtain better understandings of neuro-disease and aging with discovery of prognostic and diagnostic biomarkers and development of diagnostic techniques and medications 36 .Human brain project conducted in three phasesdescribed in Figure 2.
Figure 2.Different phases of Human Brain Proteome Project (HBPP)
Human pilot study comprises biopsy and autopsy of human brain tissues. Most participating laboratories use Protein scape platform of bioinformatics as small local database to organize and store data. This system gives benefit to gain all the data in a well-mannered and disciplinary way 34.
Matrices for Human Proteome Project:
It is basically a knowledge base for identification, quantification and characterization of protein network in broad array of biological system 34.
2013 Matrices of Human Proteome Project (HPP):
In 2013, HPP committee make matrices for whole proteome and chromosomes for protein-coding genes. In 2012, the violent estimation about “absent proteins” which means the neXtProt, PA, and GPMdb deduct from genes, is of 6568(33%) 36.
Protein evidence levels are classified into five categories which are: PEI identifies and characterize the protein way to express, identification by MS, immunohistochemistry, 3Dimensional structure and amino acid sequencing. PE2 recognize transcript expression, PE3 protein provides confirmation of similar proteins in interconnected species, PE4 provide hypothesis for gene models and PE5 contain genes that have been from same level of confirmation in past 37, 38.
2014 Matrices of Human Proteome Project (HPP):
These provides chromosomes-by-chromosomes fistulous & facilitates the work of c-HPP. NeXtProt 2014 has 1,6491 PEI entries for proteins, with 19,439 protein entries from protein existence levels 36.On new route from proteomics lab to novel proteomics, diagnostic and therapeutic in society and innovation strategy in proteomics 39. NeXtProt version 2014 was chosen as baseline for 2015 cycle from the c-HPP teams. HPP strongly agree proteome exchange of all data set and Guideline and conformation of novel findings provided by SRM and SWATH-MS methods 40, 38
2015 Matrices for Human Proteome Project (HPP):
Tissue based map of human proteome project was developed on 7 November 2014. It give the extensive annotation for cancer cell lines and drug abel etc isoforms & metabolism linked with protein based immune-histochemical studies of RNA sequencing resulted by 32 tissue 38.c-HPP workshop, EUPA annual meeting was occurred in Milan June 23-28, 41.
2016 Matrices for Human Proteome Project (HPP):
In 2016, progress made for protein knowledge between peptide Atlas and neXtProt and development of GPMBD mass spectrometry resources and human protein. NeXtProt is the primary source of knowledge related to HPP which is sourced from Swissprot or UniProtKB bases. Its mean that if data is updated by swiss-prot,is faithfully updated onto the neXtProt , maximum 3 to 4 times . Major data accumulated for PTMs of human proteome 42
Pillars of HPP:
The Biology/Disease-driven Human Proteome Project (B/D-HPP)
The main goal of the project is to study the mechanism of biological process & human diseases. It consummates by the process of research and informational tools that tell about all the protein physiology, mutation of proteins which may be reason of any disease. There is also a correlation between research method of a specific protein and research programming on that protein. Three major conclusion are drawn by the researcher 1st is major number of proteins kept as uninvestigated, 2nd is neither knowledge related to human genome nor powerful techniques of proteomics essentials and 3rd was the pattern related to research can be effected by the obtain ability of tools.18. Figure 3 and Figure 4.
Figure 3.Pillars of HPP defining different aspects of biology(Legrain, Aebersold et al. 2011)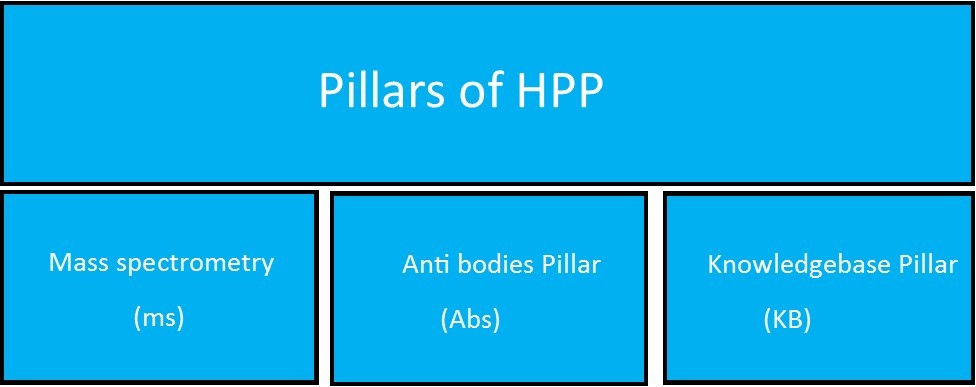
Figure 4.Components of B/D-HPPO describing involvement of different samples in human proteome project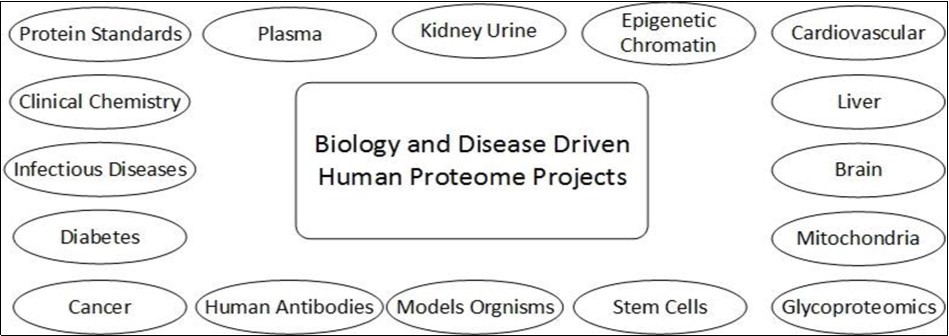
The Human Diabetes Proteome Project (HDPP):
The pathology of diabetes is the emerging issue for developing countries, the human diabetes project aim is to study better and better recognition of pathology and its all related complications. Scientific workshops and conferences are arranged maximum throughout the years to promote and share all scientific the study regarding project associated to the techniques and proposals. They are also arranged to discuss the goals of research. Different worskshops were arranged in 2013 & 2014 for the partnership and as well as other young scintist with same object in field to share their novel ideas and also findings. 22
5th HDPP Workshop in Uppsala:
The 5th HBPP in April 2014 represents the 25 top list candidates biomarkers associated with diabetes and diagnosis by plasma. 22
HPP Relation with Cancer and Biomarkers:
The first stage diagnosis of cancer is essential for its control and. Some advance approaches such as, mammograpgy and other testing provide development for the diagnosis of cancer
Improvements in technology of genomics provide quick screen for the changes in gene expression that is converted into cancerous mass of cells. Use of ELISA system to test for disease like cancer requires single confirmation of disease. High-affinity antibody that can detect the protein of interest.48. Figure 5
Figure 5.Schematic diagram of proteomic pattern diagnostics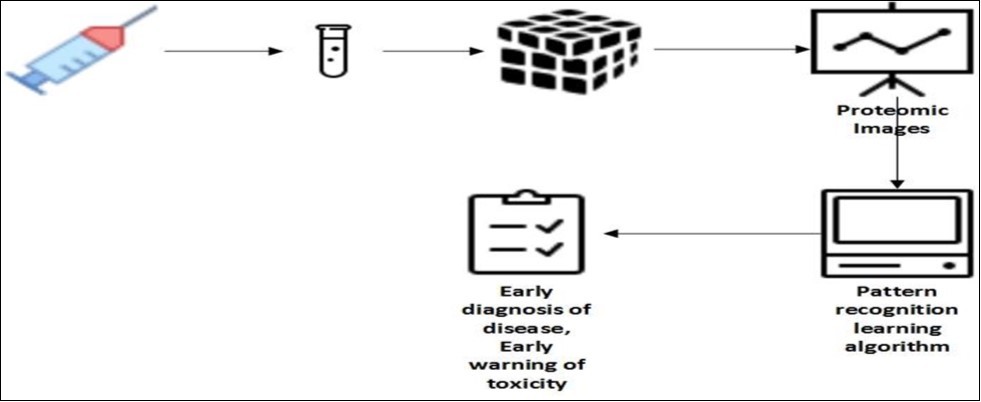
A serum sample is taken from a patient, and the proteins are attached to a chip. Mass spectrometry is implemented to achieve a proteomic image that can then be ‘read’ using bioinformatics tools. The readout can result in the early detection of cancer.
Genomic Events at the Level of Proteomics & HPP:
Genomic events & proteomics combined information typically using sequenced data base from DNA sequencing, RNA sequencing, or ribo-sequencing approaches. These research approaches is that if peptide are detected that cover all event like splicing junction non-coding RNA which is long & small ORF (open reading frame) can be improved. 1
Characterization of Post Translation Modification:
Major data resources accumulated for proteins and for the post translational modification are peptides Atlas, GPMDB,and neXtProt. The world of post translational modification is huge more than about 200 chemical classes of post translation modification are present. Peptide Atlas perform major increase in the number of observed PTMs on the base of human phosphor proteome peptide Atlas. Two different methodology are used first of all sample were searched with potential phosphorylation on the residue S, T&Y second were processed with TPP tool & PTM. It gives the possibilities that mass modification can be present on every available site. This data set is being digested with the several proteases in the laboratory of HECK & MANN laboratory in Netherland & in Munich. It demonstrated that greater possibilities for the phosphor proteomes are present when trypsin is used alone. Total 37,771 phospho peptides are identified when its hydrolyzed by the protease. And 18,000 different phospho sites are present. Regulatory mechanism is identified during experiment in which mostly tyrosine-and serine/threonine based signaling occurs. This study shows the high quantification of mitosis or signaling factor p-tyrosine is maintained at very low level when cell signaling is absent in the cell.
neXtProt have different types of modification and O-glycosylation, sumolyation, ubiquitination, nitrosylation, methylation and recently added acetylation & ADP ribosylation. In February 2016, GPMDB published new data base for the mapping of PTMs & protein modification site & genome. Protein modification are detected by the particular nucleotide variants detected by codon are basically the remapping of the splice variants is not necessary 3, 49, 50
Proteoforms and Proteins Variants:
The divergence of highly related proteins are arising at the level of cell, tissue & subcellular localization. At the DNA RNA & proteins level complexity arise by the allelic variation due to the alternative splicing due to the post translational modification. These events cause huge population of different proteins which perform many function depends on proteins nature like cell signaling inside or between the cell to regulate the gene & for the protein complex activation. For the protein analysis, two-dimensional gel electrophoresis & some new technologies are used like mass spectrometry gives a key platform for the analysis of protein complexity.
Approaches:
Two contrasting approaches are used: such as bottom up & top down approaches. In the bottom up approaches protein are digested into peptides by the use of trypsin &other proteases are also used. Then used liquid chromatography (LC) & TMS (TENDOM MASS SPECTROMETRY). In top down proteomics digestion does not occur. Proteins are direct identified by the fragmentation. In literature, one finds the different terms like proteins forms, proteins iso-forms, proteins variants, but recently proteins modified forms are used. But issue is that these all are not satisfied so that iso-from are used frequently. Functional classes of proteoforms are arising by the proteolytic cleavage & generate different proteoforms with N & C terminal. There are 1,863 peptides show that 1,703 proteoforms of 921 proteins. 2
Identification of Splice Isoforms Integration with TCGA:
RNA sequence data are rapidly accumulated clinically ,it provides opportunity to find association with the mRNA isoforms variation. Statistical methods survive for the survival analysis of mRNA isoforms variation related with patient survival time. The great strength of survive on the measurement of the uncertainty of mRNA isoforms ratio in RNA-sequence data. Survival to TCGA used for ductal carcinoma & five other types of cancer types alternative splicing is a precursor complexity of proteins.95% human genes undergoes splicing it play major role in diversity. The cancer genome Atlas (TCGA) consortium generates RNA sequence Database on the 11,000-cancer patient. Breast invasive Carcinoma (BRCA) has large size sample of RNA-sequence data over 1000 patient & information about clinical like survival time, tissue subtypes& cancer stages is available for the breast invasive carcinoma patient. This large sample size of TCGA (BRCA) data allow to cause relation between genomic & transcriptome profile to clinical outcomes & patient survival times. 51, 52
Calipho:
In 2008, after first complete manual annotation by the UniProt/Swiss-Prot group, it was believed that full set of human protein was achieved, but soon was realized that how less we know about human protein function and its characterization (PTMs, protein/protein interactions, subcellular locations, etc.). So, to gather information about what these proteins do in our body, a team was established named as CALIPHO. http://www.neXtProt anew knowledge based on human proteins. CALIPHO (Computer and Laboratory investigation of Proteins of Human Origin) is a multidisciplinary group which is carried out by the University of Geneva and the SIB, leaded by Amos Bairoch and Lydei Lane. The organization goals are: creation of software platform to integrate bioinformatics and experimental methods to determine unknown proteins and their functions, organizing data in such way that it is easy for the user to use and provide with high quality of data to the user.
To meet up these goals, CALIPHO has developed neXtProt, a human-centric protein knowledge resource. It is further working on many different experimental techniques to reveal much more about unknown proteins and their function.
Relation between CALIPHO and HPP:
About 20,300 protein-coding genes have been estimated from the analysis of the human genome. Transcriptomic analyses such as DNA microarray or RNA sequencing have manifested that these genes are expressed in a large dynamic range in the ~230 cell types that make the human body. More than fifty percent of them produces alternative splicing isoforms. During or after translation, many chemical changes of the protein products can occur (processing, post-translational modifications, etc.), resulting in a great diversity of proteoforms that differ with time, location, and physiologic or disease conditions. About one million proteoforms coexist in a single person. This variability does not take into account the inter-individual variations due to frequent polymorphisms or rare mutations. Due to recent advancement in DNA sequencing technologies, this inter-individual variability can now be examined in detail across populations. Recent progress in proteomics technologies allows detecting and quantifying proteins and their modifications with a higher accuracy. However, many proteins predicted from genomic or transcriptome analyses still are not detected, either because they were not properly evaluated, or because their expression is restricted in time and/or space, or their biophysical and chemical properties are not consistent with usual proteomics experiments. The international HUPO Human Proteome Project (HPP) was designed five years ago to try to characterize and evaluate the so-called "missing proteins" that were confidently estimated but still have not been detected at protein level. Currently, there are 2,563 such "missing proteins". To see the detailed distribution across chromosomes, go to the protein existence status, so for this purpose HPP took help of CALIPHO; i.e. neXtProt to identify these proteins. (http://www.nextprot.org/)
NeXtProt:
In the last 30 years, vast resources have been established to comprehend the molecular components and processes of human cells, for the sake of medicinal and fundamental research applications. for this purpose first target was the sequencing of the genome and the drafting of its transcriptome, it has now switched toward the study of one of the major biomolecule, the proteins. Human proteins are very complex at functional and molecular level and bioinformatics resources are needed, chiefly focused at capturing, integrating and maintaining up-to-date the available knowledge about them. 4
For this purpose, UniProt/swiss-prot groups were developed which provided us with an enormous amount of data about protein, according to estimation from the UniProtKB/Swiss-Prot knowledgebase content, 25% of these proteins (i.e. around 5000) have not been studied experimentally till now.
The data was distributed in multiple resources and websites, which caused a real problem so to solve this problem, neXtProt (http://www.nextprot.org/). All the data related to human proteome is present of a data base that is neXtProt. A common development of the SIB and of Gene Bio SA. It work like the model organism database which use for collection of data related to species and act as a one of the databest for provid stimulus/input for research on model species. As like that neXtProt provide all the data for the protein present in human 5 and to combine data correctly and develop tools that are required by the users and also provide user with high quality data and tools. 4. Figure 6.
Figure 6.NeXtProt home page having menus in header and footer making easy for the user to access all the neXtProt content, gold or silver written with the search bar refers to the quality of data.
Data Content of neXtProt:
The main data sources (as in table 1) are UniProtKB, Bgee, HPA, Peptide Atlas, SRMAtlas, GOA, dbSNP, Ensemble, COSMIC, DKFGFP-cDNA localization, Weizmann Institute of Science’s Kahn Dynamic Proteomics Database & IntAct. Other than that, for the first time ADP-ribosylation sites and new acetylation sites with their related peptides have also been loaded. With all this content, neXtProt now contains 142,453 post-translational modification sites and 1,150,170 peptides. 53. Table 2.
Table 1. brief description of pillars of HPP| MALDI-MS: | ELISA | UniProtKB |
| Used to determine the level of proteolytic processing. | used for the detection of antigen in the plasma by probing it with antibodies(Ahmad, Arya et al. 2014). | 1)The UniProt Archive (UniParc) which provides a stable, comprehensive, non‐redundant sequence collection |
| determine the presence of post-translational modifications, | 2)The UniProt Knowledgebase that give the central database for sequencesing of proteins with accuratally. | |
| also used for the analysis of smaller molecules just like peptides.(Stults 1995). | 3)The UniProt NREF databases (UniRef) provides non‐redundant data collection. (Apweiler, Bairoch et al. 2004). | |
| LC-MS/MS:used for the diagnosis of Endocrine disorders, Vitamin D analysis(Ahmad, Arya et al. 2014). | SWISS-PROTIs a data bank of accurate protein sequences, Interpretations, minimal redundancy and integration with other databases.(Junker, Contrino et al. 2000) | |
| PRIDE:There are different types of data stored in PRIDE, aim of PRIDE is to reflect the author’s analysis view on the experimental data.(Vizcaíno, Côté et al. 2012) |
| Entries | Statistics | Sources |
|---|---|---|
| Proteins/isoforms | 42196 | UniProtKB |
| Binary interactions | 192822 | IntAct |
| Post-translational modifications | 187531 | PeptideAtlas,UniProtKB,neXtProt |
| Entries with a disease | 16671 | UniProtKB |
| Entries with proteomic data | 17838 | Peptide Atlas |
| Variants | 5324509 | COSMIC,UniProtKB,dbsnp |
| Total publications | 104473 | All resources |
Human Proteome Project:
HUPO, is an international level organization which connects all the labs of proteomics which use the proteomics as a way to describe the health and mutation level of protein in the sample 54. This organization have to make the record of all proteins with respect to their existence, isoforms, variation, PTMs as well as their abundance and distribution. So the role of neXtProt within hpp combine all the result of mass spectrometry and give the matrices related to development in the project. 55
Peptide Atlas gather raw outcomes from proteomics experiments and re-explain them by using a constant informatical tool such as, the Trans-Proteomic Pipeline. Peptide Atlas provides peptide determination in biological samples 56. Same like Peptide Atlas it is also closely collaborated with UniProtKB and apply the same standard method as UniProtKB to determine protein existence. 5
Softwares and Data Accessibility:
All neXtProt annotations are available as XML and PEFF files on our FTP site (ftp://ftp.nextprot.org/). Our XML format has been modified to cope-up with the new phenotypic data. Changes are enlisted in a comment at the beginning of the new XSD file (version 2), also on the FTP site. The old XML files are no longer reachable due to technical problem. Annotations can also be obtained by our API at https://api.nextprot.org and our SPARQL endpoint (https://www.nextprot.org/proteins/. The Cellosurus – a data-base on cell lines is available at ftp://ftp.expasy.org/databases/cellosaurus/. Our software is freely reachable from the GitHub repository (https://github.com/calipho-sib) or biojs (http://www.biojs.io). 53
The neXtProt human protein knowledgebase combine data to provide comprehensive, advanced, high quality information arranged in such a way so as to present scientists around the world with a resource that make their research easier. neXtProt is continually evolving and, in terms of content, the focus will continue to be the incorporation of new variant and proteomics data in the coming future. 53
Bioinformatics Tools:
As human genome is very complex and different types of protein are also present in it and they perform different function in body. These proteins are of different kind these may functional and nonfunctional. To know about protein function, structure and disorders or mutations due to protein we need a tool or software to predict these basics. These are called bioinformatical software or tools. Table 3
Table 3. Bioinformatics tools for proteome analysis| Name of software | Function | Links |
|---|---|---|
| Phyre2 | Analysis of protein structure, function and mutation | WWW.sbg.bio.ic.ac.uk/ ~phyre/ |
| PSIPRED | Prediction of protein secondary structure | bioinf.cs.ucl.ac.uk/psipred/ |
| I-TASSER | For 3D structure and protein function annotation | Zhang lab .ccmb.med,umich.edu/I-TASSER/ |
| Dali server | Analysis of structured protein | ekhidna.biocenter.helsinki.fi/dali_server/start |
| COFACTOR | Protein function annotation | Zhang lab .ccmb.med,umich.edu/COFACTOR/ |
| JAFA server | Protein function annotation | http://jafa.burnham,org Or http://Zope.org |
| SCRATCH | Annotation of Protein structure and structural features | scratch.proteomics.ics.uci.edu |
| BLAT | Diagnose of diabetes | https://urgi.versailles.inra.fr/blat/cgi-bin/webBlat |
| Predict AD | Diagnose Alzheimer’s disease | https://www.predictad.eu/12 |
| Jpred 3 | Secondary structure prediction | www.compbio.dundee.ac.uk/jpred3 |
Software for Structure Annotation of Protein:
PSIPRED server is most reliable accurate and easy to use and developed in 2000. More than 15000 of protein structure prediction or annotation is done by this software in each month. These softwares are updated day by day due to this more reliable result are obtained.
Prediction of Secondary Structure:
PSIPRED server basically use the output of PSI -BLAST server to known the secondary structure of protein. Its accuracy to evaluate proteins secondary structure is 78 percent. 6
I-TASSER:

Dali Server:
This server is basically related to the comparative analysis of newly structured protein with ancestral protein to compare their structure sequence and function. 8
Software for Mutation and Disease Diagnosis:
Comparing DNA Sequences - Diagnosing a Rare Genetic Disease:
Basically mutation is that 1 nucleotide in1000 differ from one person to other person genom. Some variation have no affect but some have genetic mutations. As all members which carry diabetes 1 has same variation in gene that translated into insulin. By this, we find mutation and then treatment is done. 57
Predict AD project:
This software is basically use for Alzheimer disease diagnosis. The goal of this AD projects is to identify the biomarkers from different patient data for the early diagnosis and monitoring the progressive contribution by the AD project in more objective manners. 58. figure 8
Figure 8.Showing input and output of predict AD
Software for Function Annotation:
COFACTOR Software
It is basically structure based protein function annotation. Input for COFACTOR server basically require 3D structure of that particular protein whose function is necessary to be known. The output that comes by COFACTOR software is in the form of tables that show results according to our submitted proteins. 59. Figure 9
Figure 9.Shows input and output of cofactor software
JAFA Server
By giving high number of sequence of protein and structure there is need of very important and sophisticated prediction tool. In the recent few years there is vast and diverse set of software for the protein function annotation. JAFA server or software is also one of them that are used for protein structure annotation. 7
Protein Profiling
The next step is the comparison of protein profiles by the DIGE. It improves the different expression by 2-Dimensional electrophoresis, it reduce the experiment variability and allow the multivariate treatment.10, 11 This method is based on the specific labeling of proteins sample of Lys ε amino group. By the use of 3 different fluorescence probes that is cyanine’s 2, 3 and 5. These are three probes have different emission spectra and excitation without any change in protein molecular mass and isoelectric point (PI) this experiment allow the protein separation in the different sample by the use of this unique Gels which have been increase the experiment reproducibility. Number of cy dye and DIGE flour can be used
Biomedical Application:
Some common strategies are being used by different drugs to exert their effects on proteins. A particular Genetic instability is identified which cause the changes in protein structure, function and expression. Some drugs are designed to control or correct abnormalities, for eg, An inhibitor of BCR-ABL tyrosine kinas in CML is developed. CML is chronic myleogeneous leukemia. For the designing of some particular disease it is important to know about the bioactivity of protein that is important in biological processes. For example use of neutralizing antibodies & inhibitors of tyrosine kinas receptor to inhibit ontogenesis influence by the vascular endothelial growth factor in tumorous cells. Proteome is important condition in which cells exposed to the any specific disease processes. Therefore a large number of proteome for each cell. According to some hypothesis driven projects carefully some specific feature are selected that provide information for particular medical condition. Proteomic advantages with genomic capabilities, as genomic sequencing projects completed by the introduction of native proteomic funding resourcefulness, and allow the approaches which based on proteomics to realize their effects or potential in biomedical field.
Cross Analysis of Particular Proteomic based Projects:
In present cross-analysis of proteome date by organ of bio-fluid have been confirmed by various platforms. By the collective analysis of data according to primary spectra with constant criteria and bioinformatics tools easily can be compared. Via cross checking of collective analysis can improve the quality of individual analysis. Expected that HPP collaboration with the human protein quantification & detection. 43
Conclusion
After success of human genome project, scientists are working on human proteome project, for protein mapping and identification, using spectrometer, antibody capturing and bioinformatics. C-HPP and B/D-HPP work together to enhance data completeness and extensiveness while B/D-HPP provides database useful for C-HPP. Hence, human proteome project (HPP) integrate whole data about human protein that can be medicinally useful to treat many diseases, by the help of bioinformatical tools and softwares for storage and analysis of data; like protein isoforms, variants produced by post translational modification and splicing. In this review article, we have overviewed different databases such as SwissProt, UniProt, PRIDE and neXtProt providing with up-to-date and high-quality data, and softwares such as I-TASSER and BLAT. These softwares and tools are being further developed to be more easy and useful for the users. In near future, new tools are even being developed with the main focus on incorporation of new variants and proteomics data.
References
- 1.Nesvizhskii A I. (2014) Proteogenomics: concepts, applications and computational strategies.Nature methods. 11(11), 1114-1125.
- 2.Huesgen P F. (2015) LysargiNase mirrors trypsin for protein C-terminal and methylation-site identification.Nature methods. 12(1), 55-58.
- 4.Lane L. (2011) eXtProt: a knowledge platform for human proteins. Nucleic acids research. 40(D1): D 76-D 83.
- 5.Gaudet P. (2015) The neXtProt knowledgebase on human proteins: current status. Nucleic acids research. 43(D1): D 764-D 770.
- 6.Bryson K. (2005) Protein structure prediction servers at University College London. Nucleic acids research33(suppl_2):. 36-38.
- 7.Friedberg I. (2006) JAFA: a protein function annotation meta-server. Nucleic acids research34(suppl_2):. 379-381.
- 8.Holm L.P.i.Rosenstrï¿ 1/2m (2010). Dali server: conservation mapping in 3D. Nucleic acids research38(suppl_2): W545-W549.
- 9.Yang J, Zhang Y. (2015) Protein Structure and Function Prediction Using I-TASSER. Current protocols in bioinformatics. 5-8.
- 10.Westermeier R, Scheibe B. (2008) Difference gel electrophoresis based on lys/cys tagging. 2D page: Sample preparation and fractionation:. 73-85.
- 11.Richard E. (2006) Quantitative analysis of mitochondrial protein expression in methylmalonic acidemia by two-dimensional difference gel electrophoresis. , Journal of proteome research 5(7), 1602-1610.
- 12.Banks R E. (2000) Proteomics: new perspectives, new biomedical opportunities. , The Lancet 356(9243), 1749-1756.
- 13.Luscombe N M. (2001) What is bioinformatics? An introduction and overview. , Yearbook of Medical Informatics1(83-100): 2.
- 14.Muglia L J, Katz M. (2010) The enigma of spontaneous preterm birth. , New England Journal of Medicine 362(6), 529-535.
- 16.Chen Y. (2015) Identification of missing proteins defined by chromosome-centric proteome project in the cytoplasmic detergent-insoluble proteins. , Journal of proteome 14(9), 3693-3709.
- 17.Aebersold R.The Biology/Disease-driven Human Proteome Project: Enabling Protein Research for the Life Sciences Community. Journal of proteome research.
- 18.Aebersold R. (2012) The biology/disease-driven human proteome project (B/D-HPP): enabling protein research for the life sciences community. , Journal of proteome research 12(1), 23-27.
- 19.Orchard S, Hermjakob H. (2007) The HUPO proteomics standards initiative—easing communication and minimizing data loss in a changing world. Briefings in bioinformatics. 9(2), 166-173.
- 20.Hardy N W, Taylor C F. (2007) A roadmap for the establishment of standard data exchange structures for metabolomics. 3(3), 243-248.
- 21.Sansone S-A. (2007) Metabolomics standards initiative: ontology working group work in progress. 3(3), 249-256.
- 22.Schvartz D. (2015) The human diabetes proteome project (HDPP): The. Translational Proteomics8: 1-7.
- 23.Omenn G S. (2007) The HUPO human plasma proteome project. PROTEOMICS-Clinical Applications1(8):. 769-779.
- 24.M J Piccart-Gebhart. (2005) Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. , New England Journal of Medicine 353(16), 1659-1672.
- 25.Gerlinger M. (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. , New England Journal of Medicine 366(10), 883-892.
- 26.Yang J. (2012) Serum peptidome profiling in patients with gastric cancer. Clinical and experimental medicine. 12(2), 79-87.
- 27.Zhou L. (2016) Clinical proteomics-driven precision medicine for targeted cancer therapy: current overview and future perspectives. Expert review of proteomics13(4):. 367-381.
- 28.Nanjappa V. (2013) Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic acids research42(D1):. 959-965.
- 29.He F. (2005) Human Liver Proteome Project Plan, Progress, and Perspectives. Molecular & Cellular Proteomics. 4(12), 1841-1848.
- 30.Yáñez-Mó M. (2015) Biological properties of extracellular vesicles and their physiological functions. , Journal of extracellular vesicles 4(1), 27066.
- 31.Zheng J. (2006) The human liver proteome project (hlpp) workshop during the 4th hupo world congress. 6(6), 1716-1718.
- 32.Gao X. (2010) . The 2009 Human Liver Proteome Project (HLPP) Workshop , Toronto, Canada 26, 3058-3061.
- 33.Mato J M. (2007) . The 2006 Human Liver Proteome Project (HLPP) Workshops. PROTEOMICS-Clinical Applications1(5): 442-445.
- 34.Blüggel M. (2004) . Towards data management of the HUPO Human Brain Proteome Project pilot phase. Proteomics4(8): 2361-2362.
- 35.Hamacher M, Meyer H E. (2005) HUPO Brain Proteome Project: aims and needs in proteomics. , Expert review of 2(1), 1-3.
- 36.Omenn G S. (2014) A new class of protein cancer biomarker candidates: differentially expressed splice variants of ERBB2 (HER2/neu) and ERBB1 (EGFR) in breast cancer cell lines. , Journal of proteomics 107, 103-112.
- 37.Marko-Varga G.A First Step Toward Completion of a Genome-Wide Characterization of the HumanProteome.ACS.Publications.
- 38.Omenn G S. (2015) Metrics for the Human Proteome Project 2015: progress on the human proteome and guidelines for high-confidence protein identification. , Journal of proteome research 14(9), 3452-3460.
- 39.Reddy P J. (2015) The quest of the human proteome and the missing proteins: Digging deeper. Omics: a journal of integrative biology19(5):. 276-282.
- 40.Lane L. (2013) Metrics for the Human Proteome Project 2013–2014 and strategies for finding missing proteins. , Journal of proteome research 13(1), 15-20.
- 41.Zeiler M. (2012) A Protein Epitope Signature Tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Molecular and Cellular Proteomics11(3):. 111-009613.
- 42.Omenn G S. (2016) Metrics for the Human Proteome Project 2016: progress on identifying and characterizing the human proteome, including post-translational modifications. , Journal of proteome research 15(11), 3951-3960.
- 43.Legrain P. (2011) The human proteome project: current state and future direction. Molecular & cellular proteomics10(7):. 111-009993.
- 44.Ahmad Y. (2014) . Proteomics in Diagnosis: Past, Present and Future. Journal of Proteomics and Genomics1(1): 1.
- 45.Apweiler R. (2004) UniProt: the universal protein knowledgebase. Nucleic acids research32(suppl_1): D115-D119.
- 46.Junker V. (2000) The role SWISS-PROT and TrEMBL play in the genome research environment. , Journal of biotechnology 78(3), 221-234.
- 47.Vizcaíno J A. (2012) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic acids research41(D1): D1063-D1069.
- 48.Wulfkuhle J D. (2003) Proteomic applications for the early detection of cancer. , Nature reviews.Cancer 3(4), 267.
- 49.Giansanti P. (2015) An augmented multiple-protease-based human phosphopeptide atlas. Cell reports. 11(11), 1834-1843.
- 50.Sharma K. (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell reports. 8(5), 1583-1594.
- 51.Pan Q. (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing.Nature genetics. 40(12), 1413-1415.
- 52.Koonin E V, Wolf Y I. (2010) Constraints and plasticity in genome and molecular-phenome evolution. , Nature Reviews Genetics 11(7), 487-498.
- 53.Gaudet P. (2017) The neXtProt knowledgebase on human proteins:. 2017update. Nucleic acids research. 45(D1): D 177-D 182.
- 54.Paik Y-K. (2013) Genome-wide proteomics, Chromosome-Centric Human Proteome Project (C-HPP), part II,ACS,Publications.
- 55.Gaudet P. (2012) neXtProt: organizing protein knowledge in the context of human proteome projects. , Journal of proteome research 12(1), 293-298.
- 56.Farrah T. (2013) State of the human proteome in 2013 as viewed through PeptideAtlas: comparing the kidney, urine, and plasma proteomes for the biology-and disease-driven Human Proteome Project. , Journal of proteome research 13(1), 60-75.
- 57.Molven A. (2008) Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. , Diabetes 57(4), 1131-1135.
Cited by (1)
This article has been cited by 1 scholarly work according to:
Citing Articles:
Journal of Proteomics and Genomics research (2018) OpenAlex